










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.109 no.2 Buenos Aires mar./abr. 2011
PRESENTACIÓN DE CASOS CLÍNICOS
Síndrome de Sturge-Weber. Presentación de un caso con manifestaciones dermatológicas mínimas
Sturge-Weber syndrome. Report of a case with poor dermatological manifestations
Dra. Clarisa Rodofilea, Dra. Susana A. Greesa, Dra. Lidia E. Vallea, Dr. Gabriel Martinob
aServicio de Dermatología.
bServicio de Neurología. Hospital General de Niños Pedro de Elizalde.
Correspondencia: Dra. Clarisa Rodofile: clari_rodofile@yahoo.com.ar
Conflicto de intereses: Ninguno que declarar.
Recibido: 2-8-10
Aceptado: 10-01-11
RESUMEN
El síndrome de Sturge Weber (SSW) es un trastorno neurocutáneo congénito que presenta malformación vascular rojo vinosa en el territorio del trigémino, manifestaciones cerebrales (afectación leptomeníngea ipsolateral, convulsiones y retardo mental) y signos oftalmológicos (malformación vascular coroidea, glaucoma). No hay evidencia convincente de que se trate de una enfermedad hereditaria. Comunicamos un caso que presentaba una pequeña malformación vascular en región frontal y párpado superior derechos, con afectación neurológica importante; sin compromiso ocular. En el SSW no siempre la magnitud de la lesión cutánea se relaciona directamente con el compromiso del sistema nervioso central, el cual puede generar graves consecuencias sobre la salud y la calidad de vida del niño, como en nuestro caso. Destacamos la importancia del conocimiento de este síndrome neurocutáneo dada la relevancia de su pesquisa precoz, solicitud de estudios, interconsultas y el necesario abordaje interdisciplinario desde el momento del diagnóstico.
Palabras clave: Malformación vascular; Convulsiones.
SUMMARY
Sturge-Weber syndrome (SSW) is a congenital neurocutaneous disorder, which presents a port wine vascular malformation that covers the territory of the trigeminal nerve, neurological manifestations (ipsilateral leptomeningeal involvement, seizures and mental retardation) and ophthalmic signs (choroidal vascular malformation, glaucoma). There is no evidence to indicate that this is an inherited disease. Our patient had a small vascular malformation in the frontal and right upper eyelid, significant neurological involvement, and no ocular involvement. In SSW, not always the magnitude of the skin lesion is directly related to the commitment of the central nervous system, which can cause serious consequences on child's health and quality of life, as noted in our case. We emphasize the importance of being awere of this neurocutaneous syndrome given the importance of early screening, additional studies, interconsultations and the necessary interdisciplinary approach from the time of diagnosis.
Key words: Vascular malformation; Seizures.
INTRODUCCIÓN
El síndrome de Sturge-Weber (SSW), es el más frecuente de los síndromes neurocutáneos. 1 Se trata de un trastorno congénito, esporádico, que incluye la asociación de malformaciones vasculares cerebrales, cutáneas y oculares caracterizadas por una mancha color vino de oporto en el área facial del nervio trigémino, malformación vascular de las leptomeninges, epilepsia, retraso mental, déficit neurológico, glaucoma y aumento de la vascularización de la retina.1-3
Afecta ambos sexos por igual, no presenta carácter hereditario, pero se han descripto casos familiares. Tiene una incidencia de 1/50 000 a 1/230 000 personas.
El diagnóstico precoz es primordial, por los estudios, interconsultas y terapéuticas a realizar; éste debe sospecharse aunque la clínica dermatológica no sea muy manifiesta.3
HISTORIA CLÍNICA
Paciente asistido en el Servicio de Neurología desde los 4 años de edad por presentar discreta paresia braquiocrural izquierda a predominio crural; ligero retraso en la adquisición de pautas neuromadurativas motrices y lingüísticas.
A los 8 años consultó por presentar síndrome convulsivo focal, de 4 minutos de duración, en hemicuerpo izquierdo (crisis versiva cefálica), que motivó la realización de registro electroencefalográfico de sueño (resultado normal) y neuroimágenes.
Los antecedentes convulsivos del paciente, en ausencia de evidencias eléctricas o radiológicas, obligaron a considerar el diagnóstico de neuroangiomatosis, observándose en un examen más detallado la presencia de malformación vascular facial.
En interconsulta con Dermatología se observó: mácula vascular rojo vinosa, de contornos definidos e irregulares, 6 cm de diámetro, ubicada en la región frontal derecha, con progresión al párpado superior homolateral (Foto 1) y, en mucosa oral, hiperplasia gingival sin alteración vascular.
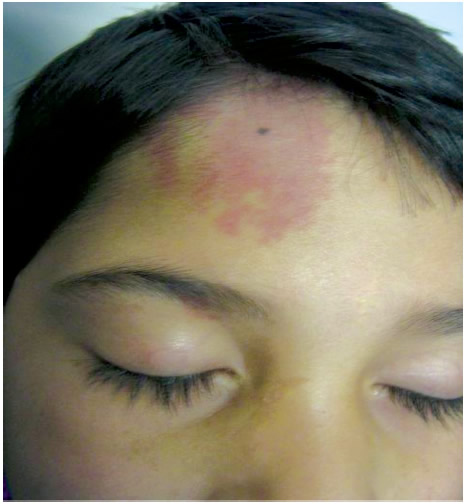
Foto 1. Mácula vascular en zona frontal con pequeña lesión de 1 x 1 cm en párpado superior derecho
La resonancia magnética nuclear (RMN) de cerebro evidenció profundización de surcos subaracnoideos, engrosamiento leptomeníngeo y atrofia cortical del hemisferio derecho, con desplazamiento de la línea media. (Foto 2)

Foto 2. RMN. Corte coronal en T2: atrofia cortical del hemisferio derecho con desplazamiento de la línea media
Las evaluaciones cardiológicas y oftalmológicas fueron normales. Un nuevo electroencefalograma objetivó discreta asimetría eléctrica de menor voltaje y mayor densidad de ondas lentas en hemisferio derecho.
Se diagnosticó: síndrome de Sturge-Weber.
COMENTARIO
El SSW es una displasia neuroectodérmica caracterizada por anomalías vasculares cerebrales, cutáneas y oculares con una expresividad variable.4
La malformación vascular capilar asociada, llamada "mancha en vino de oporto", compromete el área facial inervada por la primera rama sensitiva del nervio trigémino, particularmente el párpado superior y la zona supraorbitaria.2-3 Puede tomar las ramas maxilar y mandibular del mencionado nervio, con sobrecrecimiento óseo o de las partes blandas de la zona.2 En la evolución, la lesión tiende a oscurecerse y a adquirir zonas nodulares. Puede estar ausente en la forma "frustra" del síndrome. El 37% de los pacientes presenta lesiones bilaterales, las cuales pueden estar asociadas a compromiso intracraneal unilateral o bilateral2-4 y el 36% puede tener malformaciones vasculares en las extremidades o el tronco.3-4 Se han descripto casos de localizaciones atípicas, o sin malformación facial evidente,5 como cuero cabelludo o mucosa oral, fuera del área trigeminal, asociados a anomalía leptomeníngea.
Ante la sospecha diagnóstica es oportuno realizar interconsultas con neurología y oftalmología; asimismo, solicitar estudios por imágenes para descartar patologías asociadas.
Las malformaciones vasculares que afectan la piamadre, suelen ser ipsolaterales a la mácula cutánea y, frecuentemente, se asocian calcificaciones ipsolaterales, hemiatrofia cerebral progresiva y otras anormalidades capilares y venosas. También la alteración vascular pial puede aparecer aislada, sin lesión de coroides ni facial, en el 13% de los enfermos.6
El curso clínico de los portadores del síndrome SSW guarda una relación estrecha con las malformaciones neurológicas y los cambios neurorradiológicos, pero no con el tamaño de la lesión vascular facial; todo el hemisferio cerebral puede estar afectado.4-6
La manifestación neurológica más constante es la presencia de convulsiones, que afectan al 75- 90% de los pacientes. En el 45%, las crisis epilépticas se inician antes del año, con un pico entre los 3 y 6 meses de edad. Sólo el 7% de los pacientes inicia sus crisis después de los 5 años.7 Al principio predominan las crisis parciales motoras o tónicoclónicas generalizadas; con menos frecuencia, los espasmos infantiles, las crisis mioclónicas y atónicas. 8
Es usual la presencia de crisis frecuentes y prolongadas de difícil control farmacológico, que se logra en el 47% de los enfermos. El inicio de las crisis por debajo de los 2 años hace más probable su refractariedad;8 algunos autores refieren que las crisis epilépticas son más frecuentes e intensas cuando existe afectación bihemisférica.8
El retraso mental afecta al 50-70%8-9 de los pacientes con posterioridad al inicio de las crisis epilépticas; el 2,5% desarrolla retraso mental grave.9 Sólo el 8% de aquellos con lesiones bilaterales son intelectualmente normales.8 Las imágenes características de tomografía axial computada (TAC) cerebral o RMN, con gadolinio o sin él, pueden sugerir el diagnóstico aun antes del comienzo de las manifestaciones clínicas del SNC. La RMN ofrece mejor detalle que la TAC.10-12 Se puede observar la malformación vascular leptomeníngea que resulta en isquemia en los tejidos adyacentes, gliosis, desmielinización, calcificaciones, hemiatrofia cerebral y atrofias focales; ausencia de venas corticales superficiales adyacentes a la malformación y aumento de tamaño del sistema venoso profundo y del plexo coroideo ipsolaterales. Las calcificaciones con patrón de doble contorno que siguen las circunvoluciones cerebrales (en "vías de tren") se observan con la TAC, desde los primeros meses de vida y, con la radiografía de cráneo, desde los 7 años.10
La angiografía demuestra disminución o ausencia de venas superficiales corticales en el área de la lesión y aumento de las colaterales.
El EEG típico en estos pacientes es asimétrico, con actividad del hemisferio afectado reducida en voltaje y enlentecida.
La afectación ocular es ipsolateral a la MV; se constata glaucoma, buftalmos, malformaciones vasculares de la conjuntiva, esclerótica, coroides y retina. Con menor frecuencia, heterocromía del iris, atrofia óptica, opacificación corneal y desprendimiento de retina. Estos hallazgos no fueron observados en nuestro paciente.
En el 25% de los pacientes pueden encontrarse lesiones asociadas en la boca, principalmente alteraciones vasculares de los labios y la mucosa oral, sobrecrecimiento óseo o de partes blandas y engrosamiento gingival, como en nuestro paciente.
El diagnóstico es clínico, al observar una malformación vascular facial en el territorio del trigémino y alteraciones en SNC.
Dentro de los diagnósticos diferenciales debemos considerar: anomalía vascular aislada o asociada a síndromes: facomatosis pigmento-vascular; Parkes-Weber, Rubinstein-Taybi, síndrome PHACES, entre otros.1
La terapéutica de la epilepsia, será farmacológica o quirúrgica (o incluirá ambas). Las convulsiones responden a terapéutica anticonvulsiva en el 50% de los casos. Se recomienda el empleo de agentes antitrombóticos (aspirina) en lesiones de gran tamaño, pues disminuyen los episodios trombóticos y el deterioro progresivo.
La hemisferectomía está indicada sólo en pacientes con malformación vascular leptomeníngea extensa unilateral, con hemiparesia unilateral y convulsiones intratables.13 Vázquez y col., en su trabajo sobre hemisferectomías y hemi-hemisferectomías, manifiestan que, para el manejo de las epilepsias refractarias al tratamiento farmacológico, dichos procedimientos ofrecen un alto índice de resultados positivos con baja morbimortalidad. 14
El tratamiento del glaucoma es, en general, difícil y los pacientes requieren cirugía.
La malformación vascular facial en general representa un problema cosmético. La terapia con láser de colorante pulsado es la mejor alternativa de tratamiento estético, con bajo riesgo y dolor variable.15 Se pueden también utilizar maquillajes cubritivos.
La estimulación precoz se destaca para mejorar el retraso mental y los déficit motores.
CONCLUSIÓN
La presencia de una malformación vascular capilar en la zona trigeminal, aun pequeña, observada desde el nacimiento, debe despertar la sospecha de SSW. Este síndrome neurocutáneo, que afecta piel, SNC y ojos, exige interconsultas tempranas a neurólogos, neurocirujanos, oftalmólogos, dermatólogos y terapistas físicos. El abordaje interdisciplinario beneficia a los pacientes en su calidad de vida presente y futura. Es valioso insistir en la necesidad de la exhaustiva búsqueda de patología dermatológica o neurológica asociada; aunque en oportunidades falte la correlación.
1. Hering S, Sarmiento FGR, Valle LE. Actualización en el diagnóstico y tratamiento de los hemangiomas. Rev Argent Dermatol 2006;87(1):54-66. [ Links ]
2. Lin DDM, Gailloud P, McCarthy EF, Comi AM. Oromaxillofacial osseous abnormality in Sturge-Weber syndrome: case report and review of the literature. Am J Neuroradiol 2006;27(2):274-277. [ Links ]
3. Paller AM, Mancini AJ. Hurwitz. Clinical Pediatric Dermatology. 3ª Ed. Filadelfia: Elsevier Saunders; 2006.Págs.265- 306; 323-324. [ Links ]
4. Mirowski G, Liu A, Stone M, Caldemeyer K. Sturge-Weber syndrome. J Am Acad Dermatol 1999;41(5 pt1):772-773. [ Links ]
5. Incecik F, Herguner MO, Ozcan K, Altunbasak S. An 18-month-old girl with a history of convulsions and a facial nevus. Ann Saudi Med 2008;28(2):138. [ Links ]
6. Rodríguez-Barrionuevo AC. Síndromes neurocutáneos de anomalías vasculares. Rev Neurol 1996;24(133):1072-1084. [ Links ]
7. Sujanski E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet 1995;57(1):35-45. [ Links ]
8. Gómez MR, Bebin EM. Sturge-Weber syndrome. En: Gómez MR ed. Neurocutaneous diseases: a practical approach. Londres: Butterworths; 1987.Págs.356-367. [ Links ]
9. Roach ES. Neurocutáneos syndromes. Pediatr Clin North Am 1992;39(4):591- 617. [ Links ]
10. Pascual-Castroviejo I, Díaz González C, García Relian RM, González Casado I, Muñoz-Hiraldo E. Sturge Weber syndrome: study of 40 patients. Pediatr Neurol 1993;9(4):283- 288. [ Links ]
11. Batista CE, Chugani HT, Hu J, Haacke EM, et al. Magnetic resonance spectroscopic imaging detects abnormalities in normal-appearing frontal lobe of patients with Sturge- Weber syndrome. J Neuroimaging 2008;18(3):306-313. [ Links ]
12. Boukobza M, Enjolras O, Cambra MR, Merland JJ. Syndrome de Sturge-Weber. Données actuelles de l´imagerie neuroradiologique. J Radiol 2000;81(7):765-771. [ Links ]
13. Medina MT, Kraemer D, Solano M. Functional hemispherectomy in adult patients with catastrophic epilepsy: a new therapeutic indication? Neurología 2009;24 (1):7-8. [ Links ]
14. Vázquez C, de Jesús Barrios L, Bartuluchi M, Medina C, et al. Hemisferectomías y Hemi-Hemisferectomías: nuestra experiencia acerca de 49 casos. Rev Argent Neuroc 2008; 22(5):131-133. [ Links ]
15. Léauté-Labrèze C, Boralevi F, Pedespan J-M, Meymat Y, Taïeb A. Pulsed dye laser for Sturge-Weber syndrome. Arch Dis Child 2002;87:434-435. [ Links ]

