










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.109 no.5 Buenos Aires sept./oct. 2011
http://dx.doi.org/10.5546/aap.2011.e91
Presentación de casos clínicos Seudohipoaldosteronismo de tipo 1: una emergencia hidroelectrolítica infrecuente. Comunicación de cuatro casos
Pseudohypoaldosteronism type 1: an uncommon electrolyte emergency. Report of four cases
Dr. Gustavo Roberto Marína
a. Unidad de Nefrología. Hospital de Niños "Héctor Quintana". Jujuy, Argentina.
Correspondencia: Dr. Gustavo Roberto Marín: tama@imagine.com.ar Conflicto de intereses: Ninguno que declarar.
Recibido: 17-2-11
Aceptado: 5-4-11
RESUMEN El seudohipoaldosteronismo de tipo 1 es un síndrome infrecuente de resistencia a la aldosterona que se manifiesta con pérdida salina, hiponatremia, hiperpotasemia, acidosis metabólica hiperclorémica e hiperaldosteronismo hiperreninémico. El síndrome puede ser genético; secundario a uropatías e infección urinaria entre otras causas o presentarse esporádicamente. La pérdida salina puede ser sistémica y grave o localizada a nivel renal, por lo general, con mejor pronóstico. El cuadro clínico se manifiesta predominantemente en los primeros siete meses de vida; un marcado retraso pondoestatural y vómitos recurrentes suelen ser los signos clínicos habituales, rara vez se presenta como una emergencia hidroelectrolítica en forma de shock hipovolémico, arritmias cardíacas hiperpotasémicas y crisis convulsiva por hiponatremia. Se presentan cuatro pacientes que debutaron como una emergencia hidroelectrolítica.
Palabras clave: Seudohipoaldosteronismo de tipo 1; Hiponatremia; Hiperpotasemia; Aldosterona; Infección urinaria.
SUMMARY
Pseudohypoaldosteronism type 1 is a rare syndrome of resistance to aldosterone manifested by salt wasting, hyponatremia, hyperkalemia, hyperchloremic metabolic acidosis, and hiperreninemic hyperaldosteronism. The syndrome may be genetic, secondary to uropathies and urinary tract infection among other causes or it may occur sporadically. The salt wasting may be systemic and severe or localized to the kidney usually with better prognosis. The clinical picture is prevalent in the first seven months of life, failure to thrive and recurrent vomiting are usually the common clinical signs, an electrolyte emergency in the form of hypovolemic shock, hyperkalemic cardiac arrhythmias and hyponatremic seizures is rare. Four patients presenting with an electrolyte emergency are reported
Key words: Pseudohypoaldosteronism type 1; Hyponatremia; Hyperkalemia; Aldosterone; Urinary tract infection.
doi:10.5546/aap.2011.e91
INTRODUCCIÓN
La aldosterona es un mineralocorticoide que actúa principalmente en el riñón y secundariamente en colon, pulmón y glándulas sudoríparas, salivales y lagrimales. Su acción requiere, a nivel celular, la existencia del receptor mineralocorticoide (RM) y de proteínas transportadoras de sodio denominadas canales epiteliales de sodio (CES), que actúan a nivel apical, donde favorece la reabsorción electrogénica de sodio y la eliminación de potasio hacia la orina.1,2
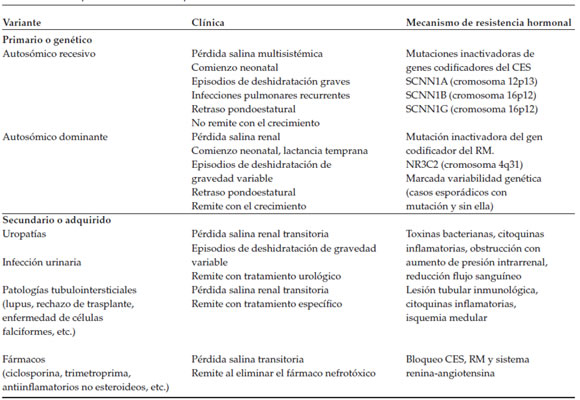
El seudohipoaldosteronismo de tipo 1 (PH1) es un síndrome de resistencia mineralocorticoide infrecuente que se caracteriza por pérdida salina, hiponatremia, hiperpotasemia, acidosis metabólica hiperclorémica e hiperaldosteronismo hiperreninémico. Se han identificado formas genéticas autosómicas recesivas (AR) y dominantes (AD), casos esporádicos y un tipo secundario a uropatías e infección urinaria, entre otras causas (Tabla 1).1-7 Por lo general, el síndrome se presenta de manera insidiosa, rara vez se manifiesta como una emergencia hidroelectrolítica como lo ejemplifican los casos que se presentan, tres de tipo secundario y uno genético AD.
Tabla 1. Seudohipoaldosteronismo de tipo 1
A continuación, se describe el síndrome clínico de cada paciente y en la Tabla 2 figuran los datos de laboratorio y estudios radiológicos efectuados. Los pacientes comparten tres aspectos clínicos: un cuadro de deshidratación variable, un trastorno electrolítico similar y un volumen urinario diario inapropiadamente alto para el grado de deshidratación: 3,6-6,7 cm3/kg/h (poliuria).
Tabla 2. Estudios de laboratorio y radiológicos

Caso 1 Niño de 2 meses de vida que consulta por vómitos y diarrea moderada de 48 h de evolución, en sala de emergencias presenta una convulsión generalizada que cede con diazepam. Ingresa con deshidratación moderada y un peso de 2900 g (3000 g al nacer). Se indicó cloruro de sodio al 3% y bicarbonato sódico parenteral. Se continuó la hidratación por vía oral con evolución favorable. El aporte de sodio oral llegó a picos de 22 mmol/ kg/día, con normalización de la natremia un mes después. A los 15 meses su natremia fue de 141 mmol/l, potasemia 5,9 mmol/l y bicarbonatemia 18 mmol/l, peso y talla entre percentilos 25-50 sin aportes de sodio ni de mineralocorticoides. Sus antecedentes familiares revelaron padres no consanguíneos, adolescentes, madre sana, sin datos paternos. La madre del niño tiene 13 hermanos: 6 mujeres (3 fallecidas antes del año de vida -causa desconocida- y 3 sanas) y 7 varones (todos fallecidos, 4 antes del año de vida y 3 entre el año y los dos años, dos diagnosticados como "hiperplasia suprarrenal congénita"). Los padres no regresaron para estudio hormonal.
Caso 2 Niña de 7 meses de vida portadora de anomalía de tipo VACTERL (ano imperforado; cardiopatía congénita, escoliosis dorsolumbar, vértebras lumbares en cuña; agenesia radial derecha; hidronefrosis bilateral; doble uretra incompleta de tipo 1); colostomizada; que consulta por gastroenteritis aguda febril de 3 días de evolución. Ingresa en shock hipovolémico con un peso de 4550 g (al nacer 4130 g). Se realizan tres expansiones con solución fisiológica, aporte de bicarbonato sódico parenteral y cefalotina. Continúa con sales de hidratación oral y normaliza sus electrólitos en 72 h. Al año de vida, peso en percentilo 25 con electrólitos normales. Se hace reimplante ureteral bilateral.
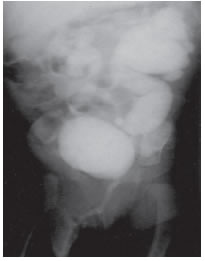
Figura 1. Caso 2. Cistouretrografía miccional. Hidronefrosis bilateral. Reflujo vesicoureteral grado 5 izquierdo, grado 4 derecho.
Caso 3 Niña de 1 mes de vida con gastroenteritis aguda febril de 48 h de evolución. Ingresa en shock hipovolémico con un peso de 2700 g (al nacer 2780 g), el examen físico evidencia clítoris prominente, seno urogenital y un tumor abdominal. Se realizan dos expansiones con solución fisiológica, aporte de bicarbonato sódico y cefalotina. Continúa con sales de hidratación oral y normaliza electrólitos al cuarto día. Dos meses después reitera episodios de infección urinaria sin alteraciones electrolíticas. Se realiza vesicostomía descompresiva. Peso al año de vida en percentilo 50. Cirugía correctora pendiente.

Figura 2. Caso 3. Tomografía computada abdominal. Hidrocolpos tabicado con desplazamiento y compresión vesical anterior
Caso 4 Niña de 2 meses de vida con vómitos recurrentes de 72 h de evolución, con la consulta presenta una convulsión afebril generalizada. Ingresa en shock hipovolémico y signos electrocardiográficos de hiperpotasemia, el peso de ingreso es de 3710 g (al nacer 3200 g). Recibe dos expansiones con solución fisiológica, cloruro de sodio al 3%, bicarbonato sódico, gluconato cálcico al 10%, nebulizaciones con salbutamol y cefotaxima. Se continúa con hidratación oral y normaliza electrólitos en 72 h. Al mes del egreso hospitalario presenta nueva infección urinaria con similar trastorno hidroelectrolítico sin convulsiones. No concurre a controles posteriores.
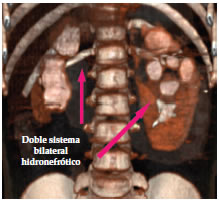
Figura 3. Caso 4. Tomografía computada abdominal. Urograma excretor tridimensional. Doble sistema bilateral hidronefrótico. Atrofia y displasia renal derecha. Displasia polar superior izquierda
DISCUSIÓN
Un marcado retraso pondoestatural y vómitos recurrentes suelen ser las manifestaciones clínicas habituales del PH1; otras incluyen diarrea, brotes de deshidratación, constipación, poliuria que suele pasar desapercibida y fiebre, en caso de infección urinaria (puede estar ausente -caso 4-). El síndrome completo y grave suele expresarse en los primeros siete meses de vida por la inmadurez tubular y baja ingesta de sodio con la lactancia, aunque puede haber diagnósticos tardíos;4-7 en la segunda infancia, los casos con pielonefritis pueden cursar solo con índices de excreción de potasio alterados y aumento hormonal.8 El cuadro clínico agudo de un PHA1 puede resultar en una amenaza para la vida del paciente,4,7,9,10 lo cual no es habitual dada la presentación solapada del síndrome. En este trabajo, el colapso circulatorio, una hiperpotasemia sintomática y un edema cerebral por hiponatremia aguda fueron las manifestaciones de emergencia. Una sepsis secundaria a foco urinario también debe considerarse como riesgo potencial; por lo tanto, el síndrome requiere un reconocimiento y tratamiento precoces para estabilizar al paciente. Un plan de hidratación adecuado normaliza el trastorno electrolítico en alrededor de 72 h; la hidratación oral, si es tolerada, es particularmente útil para el manejo lento de una hiponatremia crónica asintomática. El tratamiento crónico en los tipos genéticos y esporádicos consiste en administrar sales de sodio a largo plazo para evitar un trastorno del crecimiento; la pérdida salina disminuye hacia la segunda infancia con la maduración absortiva tubular proximal, del sistema renina-angiotensina-aldosterona y mayor ingesta salina con la dieta.3,5-8 Ante una hiperpotasemia resistente al tratamiento, pueden ser útiles las resinas de intercambio catiónico.
En los casos secundarios, aunque el cuadro clínico remita rápidamente con el tratamiento, la resistencia hormonal puede persistir por un tiempo variable y existe riesgo de retraso de crecimiento, lo cual debe evaluarse periódicamente; una cirugía urológica precoz reducirá o eliminará el impacto negativo sobre la función renal. El aporte de mineralocorticoides exógenos en los diferentes tipos de PH1 no tiene efecto alguno, por lo que no están indicados.1-7,9-11 En neonatos y lactantes, los diagnósticos diferenciales por considerar ante la presencia de hiponatremia, hiperpotasemia, acidosis metabólica y natriuria aumentada se detallan en la Tabla 3.7,9-11,15
Tabla 3. Seudohipoaldosteronismo de tipo 1: diagnósticos diferenciales
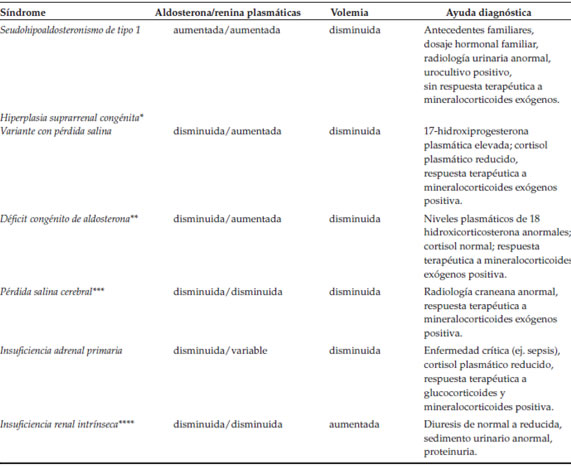
* Citas bibliograficas 6, 7, 9, 10.
** Cita bibliografica 11.
*** Debe considerarse este diagnostico ante la presencia de sintomas neurologicos.
**** Inicialmente, el fallo renal en el PH1 es habitualmente prerrenal y poliurico -por sobrecarga osmotica y deficit de concentracion (uropatias)- en lugar de oligurico para el grado de deshidratacion (citas bibliograficas 5-7, 9, 10, 15).
Este trabajo además evidencia la heterogeneidad clínico-fenotípica del síndrome. Tres pacientes se presentaron con anomalías urológicas y en dos de ellos asociadas con síndromes malformativos: uno con cloaca posterior (un caso similar fue descripto previamente)14 y otro con anomalía VACTERL (sin asociación previa descripta). Si bien no hay datos epidemiológicos sobre la incidencia relativa de los diferentes tipos del PH1, quizás, el secundario a patología urológica e infección urinaria sea el más común, aunque subdiagnosticado, por la frecuencia significativa de estos trastornos en pediatría.5 En el 80% de los casos se asocia con uropatías unilaterales o bilaterales con obstrucción o sin ella; uropatías sin infección urinaria y con infección urinaria aislada (menos del 5% de los casos), rara vez existe displasia renal asociada. Los menores de tres meses representan el 90% de los casos.5,6 El caso 1 es probablemente un PH1 autosómico dominante, por los antecedentes familiares, la evolución clínica y la recuperación electrolítica parcial. Este tipo tiene una expresión fenotípica altamente variable; la presentación clínica oscila desde portadores asintomáticos con alteración hormonal o sin ella hasta un síndrome completo grave lo cual evidencia que esta forma genética puede tener un curso no siempre favorable como suele describirse.12,13
En conclusión, el PH1 puede presentarse insidiosamente, con retraso pondoestatural o como una emergencia hidroelectrolítica en el neonato y el lactante. Debe ser incluido entre los diagnósticos diferenciales de una crisis convulsiva y cuadros gastrointestinales con marcada hiponatremia e hiperpotasemia. El examen de orina y los estudios por imágenes urinarios pueden contribuir a descubrir una uropatía asociada.
1. Riepe FG. Clinical and molecular features of type 1 pseudohypoaldosteronism. Horm Res 2009;72(1):1-9. [ Links ]
2. Bonny O, Rossier BC. Disturbances of Na/K balance: pseudohypoaldosteronism revisited. J Am Soc Nephrol 2002;13(9):2399-414. [ Links ]
3. Geller DS. Mineralocorticoid resistance. Clin Endocrinol (Oxf) 2005;62(5):513-20. [ Links ]
4. Viemann M, Peter M, López-Siguero P, Simic-Schleicher G, Sippell WG. Evidence for genetic heterogeneity of pseudohypoaldosteronism type 1: identification of a novel mutation in the human mineralocorticoid receptor in one sporadic case and no mutations in two autosomal dominant kindreds. J Clin Endocrinol Metab 2001;86(5):2056-209. [ Links ]
5. Belot A, Ranchin B, Fichtner C, Pujo L, et al. Pseudohypoaldosteronisms, report on a 10-patient series. Nephrol Dial Transplant 2008;23(5):1636-41. [ Links ]
6. Bogdanovic R, Stajic N, Putnik J, Paripovic A. Transient type 1 pseudo-hypoaldosteronism: report on an eight-patient series and literature review. Pediatr Nephrol 2009;24(11): 2167-75. [ Links ]
7. Dillon M, Leonard JV, Buckler JM, Ogilvie D, et al. Pseudohypoaldosteronism. Arch Dis Child 1980;55(6):427-34. [ Links ]
8. Rodríguez-Soriano J, Vallo A, Quintela M, Oliveros R, Ubetagoyena M. Normokalaemic pseudohypoaldosteronism is present in children with acute pyelonephritis. Acta Paediatr 1992;81(5):402-6. [ Links ]
9. Thies K, Boos K, Muller-Deile K, Ohrdorf W, et al. Ventricular flutter in a neonate-severe electrolyte imbalance caused by urinary tract infection in the presence of urinary tract malformation. J Emerg Med 2000;18(1):47-50. [ Links ]
10. Mastrandrea L, Martin D, Springate J. Clinical and biochemical similarities between reflux/obstructive uropathy and salt-wasting congenital adrenal hyperplasia. Clin Pediatr (Phila) 2005;44(9):809-12. [ Links ]
11. Giapros V, Tsatsoulis A, Drougia E, Kollios K, et al. Rare causes of acute hyperkalemia in the 1st week of life. Three case reports. Pediatr Nephrol 2004;19(9):1046-49. [ Links ]
12. Chitayat D, Spirer Z, Ayalon D, Golander A. Pseudohypoaldosteronism in a female infant and her family: diversity of clinical expression and mode of inheritance. Acta Paediatr Scand 1985;74(4):619-22. [ Links ]
13. Geller D, Zhang J, Zennaro M, Vallo-Boado A, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol 2006;17(5): 1429-36. [ Links ]
14. Iliev D, Petruch U, Ranke M, Binder G, et al. Transient pseudohypoaldosteronism with complex malformation of internal genitalia. A case report. Horm Res 2000;54(3): 149-52. [ Links ]
15. Chandar J, Abitbol C, Zilleruelo G, Gosalbez R, et al. Renal tubular abnormalities in infants with hydronephrosis. J Urology 1996;155(2):660-3. [ Links ]

