










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.110 no.4 Buenos Aires jul./ago. 2012
COMUNICACIÓN BREVE
Terapia de reemplazo enzimático en la forma infantil de la enfermedad de Pompe: experiencia de un caso con 7 años de seguimiento en Argentina
Enzyme replacement therapy in the infantile form of Pompe disease: Argentinean experience in a seven-year follow up case
Dr. Hernán M. Amartinoa y Dr. Brian M. Cavagnaria
a. Departamento de Pediatría. Hospital Alemán. Ciudad Autónoma de Buenos Aires, Argentina.
Correspondencia: Dr. Hernán Amartino: hernan.amartino@gmail.com
Conflicto de intereses: Ninguno que declarar.
Recibido: 21-12-2011
Aceptado: 4-5-2012
http://dx.doi.org/10.5546/aap.2012.323
RESUMEN
La forma infantil de la enfermedad de Pompe conduce al óbito antes del año de vida por miocardiopatía o insuficiencia ventilatoria. Presentamos la experiencia de siete años de terapia de reemplazo enzimático en un niño diagnosticado a los 7 días de vida; se trata del seguimiento más prolongado en el país. El tratamiento fue bien tolerado, sin reacciones adversas asociadas. Los parámetros ecocardiográficos y electrocardiográficos se normalizaron progresivamente en el primer año y se mantuvieron estables. El niño logró rolar y sentarse sin sostén, pautas que se perdieron a partir del tercer año. Ingresó en respirador a los 16 meses. Se mantiene vivo con 7 años de edad, con debilidad muscular generalizada grave. El niño superó notablemente la edad promedio de sobrevida y de ingreso a respirador. Fue clara la mejoría cardíaca, pero el beneficio sobre el músculo esquelético fue limitado.
Palabras clave: Enfermedad de Pompe; Terapia de reemplazo enzimático; Alfa-glucosidasa ácida; Argentina; Myozyme®.
SUMMARY
The infantile form of Pompe disease drives children to death before the first year of life due to cardiomyopathy and respiratory insufficiency. We present the seven-year follow-up experience with enzyme replacement therapy on a child with Pompe disease, being the longest follow-up in the country. The treatment was well tolerated without adverse reactions. The echocardiographic and electrocardiographic parameters clearly improved during the first year and remain stable. Motor milestones (like rolling over or sitting down without support) were initially achieved, but, after the third year were getting lost. The average age of ventilator dependence was also delayed (16 months). The 7-year old patient remains alive with severe generalized muscle weakness. The child notably overcame the average age of survival and onset of ventilator dependence. Although the cardiovascular improvement was clear, enzyme replacement therapy efficacy on skeletal muscle was limited in this patient.
Key words: Pompe disease; Enzyme replacement therapy; Acid alphaglucosidase; Argentina; Myozyme®.
INTRODUCCIÓN
La enfermedad de Pompe tiene una incidencia global de 1 en 40 0001 y se hereda de forma autosómica recesiva. Se genera por el déficit de la enzima lisosomal alfa-glucosidasa ácida, que hace que el glucógeno lisosomal se acumule anormalmente, lo cual afecta a los elementos contráctiles de las fibras del músculo esquelético y cardíaco.2
La forma infantil de la enfermedad de Pompe se caracteriza por hipotonía, debilidad muscular y miocardiopatía, aunque también se aprecia dificultad para alimentarse, retraso motor, hepatomegalia y dificultad respiratoria. Generalmente, estos niños mueren antes del año de vida3,4 por fallo cardiorrespiratorio.5
El mayor avance en el tratamiento de la enfermedad de Pompe fue el desarrollo de la terapia de reemplazo enzimático, que consiste en realizar infusiones periódicas de alfa-glucosidasa ácida humana recombinante.
El objetivo del presente artículo es presentar la experiencia de siete años de seguimiento de un niño con enfermedad de Pompe infantil, en tratamiento con terapia de reemplazo enzimático.
CASO CLÍNICO
Primer hijo de padres no cosanguíneos, nacido de término. A los 5 días presentó cianosis peribucal, palidez, hipotonía y bradicardia, por lo que ingresó a cuidados intensivos neonatales. El electrocardiograma presentó intervalo PR acortado y complejos QRS gigantes. El ecocardiograma mostró miocardiopatía hipertrófica no obstructiva, septum interventricular hipertrófico y disfunción diastólica. El índice de masa del ventrículo izquierdo fue de 212,9 g/m2 (percentilo 97= 86,6 g/m2). Presentó transaminasas elevadas y una CPK inicial de 1576 UI/l (VN menor a 295 UI/l).
Ante la posibilidad diagnóstica de enfermedad de Pompe,6 se midió el cociente alfa-glucosidasa neutra/ácida y la actividad enzimática en linfocitos, para finalmente confirmar el diagnóstico de forma infantil de la enfermedad de Pompe por biología molecular (Tabla 1).
Tabla 1. Resultados bioquímicos y moleculares

a Cociente α-glucosidasa lisosomal neutra/ácida en GSPF (gotas de sangre en papel de filtro): Normal <28,8.
b Actividad de α-glucosidasa ácida en linfocitos: Pompe: 0,0-0,07 nmol/mg proteína/min. Normal: 0,49-1,27 nmol/mg proteína/min.
c CPK: creatín fosfokinasa. Valor normal adultos < 190 UI/l; niños < 295 UI/l.
d Nomenclatura de mutaciones según www.hgvs.org/mutnomen/
Se estudiaron ambos progenitores, hallándose que el niño había heredado la mutación c.1210G>A (p.Asp404Asn) de su padre y la mutación c.2432Del_T (p.Leu811_fs36X) de su madre, situación que comunicamos previamente.7
Con 45 días de vida, el niño presentaba macroglosia, hipotonía muscular generalizada y arreflexia. A los dos meses y medio mostraba debilidad de miembros, la cual alcanzó al tronco un mes más tarde. Con cuatro meses, presentaba hepatomegalia y retraso madurativo, sin lograr el sostén cefálico.
A los 5 meses de edad ingresó en un programa de tratamiento compasivo con alfa-glucosidasa ácida humana recombinante (Myozyme®, Genzyme Corp.), administrada en infusión endovenosa (20 mg/kg/dosis) cada 14 días. Desde los 5 años de vida se duplicó la dosis (20 mg/kg/dosis cada 7 días). Actualmente continúa con este tratamiento.
Los resultados obtenidos a lo largo de siete años de seguimiento fueron los siguientes:
Aspectos generales del tratamiento y expectativa de vida
El niño, de 7 años de edad, continúa con vida. La terapia de reemplazo enzimático fue bien tolerada, sin reacciones adversas asociadas.
Aspecto cardiovascular
La hepatomegalia desapareció a los 2 meses de tratamiento. Mejoraron todos los parámetros ecocardiográficos y electrocardiográficos, con una disminución de la hipertrofia del septum interventricular y del índice de masa del ventrículo izquierdo. Este último, que al momento del diagnóstico era mayor a 200 g/m2, fue descendiendo progresivamente durante la terapia de reemplazo enzimático. A los 12 meses alcanzó un valor por debajo del máximo normal para la edad y hasta el día de hoy nunca retrogradó.
Actualmente exhibe sólo una ligera hipertrofia del septum interventricular, con buena función ventricular y doppler cardíaco normal. No presenta síntomas cardiológicos ni recibe medicación cardiovascular. Continúa con seguimiento electrocardiográfico y ecocardiográfico semestral.
Aspecto respiratorio
Presentó una disfunción lentamente evolutiva de su mecánica ventilatoria. A los 15 meses fue hospitalizado con insuficiencia respiratoria aguda secundaria a neumonía aspirativa. A los 16 meses requirió una traqueostomía con asistencia respiratoria mecánica inicialmente nocturna y, luego de 6 meses, permanente. Actualmente recibe broncodilatadores, corticoides inhalados y tratamiento kinesiológico diario.
Aspecto nutricional
A los 16 meses se le realizó una funduplicatura gástrica con gastrostomía, por la cual se alimenta con una dieta hipercalórica (superando el 10-15% del requerimiento calórico para la edad), con un aporte proteico del 20%. Presenta progreso de peso adecuado, manteniéndose entre los percentilos 50 y 75 para su edad.
Aspecto neurológico y madurativo
Al inicio de la terapia de reemplazo enzimático, con 5 meses, el niño apenas sostenía su cabeza de forma estable (Figura 1). En el primer año logró pautas madurativas como rolar, adquirir el sostén cefálico (6,3 meses) y sentarse sin apoyo (9,5 meses) (Figura 2), aunque nunca logró mantenerse en pie. Logró manipular objetos con sus manos. Sin embargo, a los 2 años comenzó a aumentar la debilidad generalizada, primordialmente a nivel axial y proximal de miembros (cintura pelviana, tronco y cuello). A los 2 años y medio perdió la sedestación independiente. Fue notoria la regresión de sus habilidades motrices gruesas y finas, incluso a nivel distal, con menor fuerza de prensión manual. Por esta insuficiente respuesta a la terapia de reemplazo enzimático a dosis habituales, desde los 5 años se duplicó la frecuencia sin obtener hasta el momento mejorías significativas. Actualmente está en cama, sin posibilidad de vencer la gravedad con el cuello ni con los miembros (Figura 3). Realiza estimulación temprana y kinesiología motora.

Figura 1. Paciente con enfermedad de Pompe infantil en terapia de reemplazo enzimático a los 5 meses. Hipotonía axial con sostén cefálico incompleto. Nótese la sonda nasogástrica para incrementar el peso
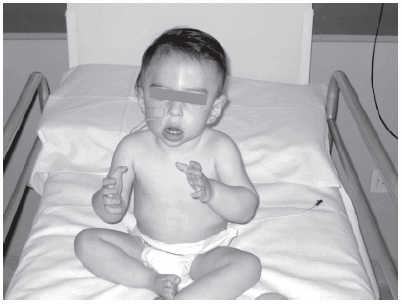
Figura 2. Paciente con enfermedad de Pompe infantil en terapia de reemplazo enzimático a los 11 meses. Logra la sedestación sin apoyo, con buen control del tronco. Tiene buen uso de los miembros superiores. Leve hipotonía facial que otorga facies característica
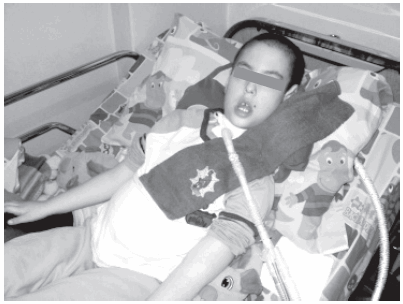
Figura 3. Paciente con enfermedad de Pompe infantil en terapia de reemplazo enzimático a los 6 años. En cama con poca posibilidad de vencer la gravedad
La clasificación de la función motora gruesa GMFCS-E & R8 considera el movimiento autoiniciado por el paciente, no tanto en función de la calidad del movimiento sino en relación a los cambios que resulten significativos para la vida diaria (limitaciones funcionales, uso de dispositivos auxiliares de la marcha, etc.). Se definen cinco niveles, y el V es el de mayor dependencia. En nuestro caso, el niño presentó un nivel III hasta los 2 años de edad. A partir de los 3 años y medio y hasta la actualidad, se encuentra en nivel V.
En el aspecto cognitivo, a los 14 meses mostraba un índice de desarrollo normal (test de Bayley) con retraso en la expresión del lenguaje. Durante la evolución mostró niveles de inteligencia no verbal acordes a su edad. El retraso en el lenguaje expresivo fue secundario a la discapacidad motora fono-articulatoria.
Desde los cuatro años orina por rebosamiento.
Aspecto inmunológico
El título de anticuerpos contra la enzima exógena (IgG anti alfa-glucosidasa ácida humana recombinante) se elevó a partir de los 5 meses (título= 100) y alcanzó un máximo entre los 6 y 8 meses (título= 400). Tras esta seroconversión inicial, el título se negativizó a los 9 meses (inmunotolerancia).
Aspecto social
El niño se encuentra bien conectado. No logra vocalizar lenguaje verbal pero emite sonidos con sentido. Su lenguaje gestual está limitado por su debilidad muscular, pero se comunica asintiendo o negando con movimientos oculares.
DISCUSIÓN
Un 92-95% de los niños con enfermedad de Pompe infantil muere durante el primer año de vida (media entre 6 y 8,7 meses).9,10 Como contrapartida, nuestro paciente superó los 7 años de vida, transformándose en el caso de seguimiento más prolongado del país.
Al día de hoy, la terapia de reemplazo enzimático es la única terapia específica y la mejor alternativa posible para el tratamiento de los niños con enfermedad de Pompe. Con siete años de seguimiento, nuestro paciente no presentó efectos adversos relacionados con la terapia de reemplazo enzimático, ni siquiera al duplicar la dosis.
La terapia de reemplazo enzimático demostró su capacidad para revertir la patología del músculo cardíaco: disminuyó la cardiomegalia,11 mejoró la función ventricular12,13 y las anomalías de conducción.13,14 Hallamos una disminución de la hipertrofia del septum interventricular, normalización del índice de masa del ventrículo izquierdo al año de tratamiento y recuperación de la función ventricular, sin involución hasta la fecha.
El otro eje central de la enfermedad de Pompe es la debilidad muscular progresiva que determina un lactante hipotónico y que lleva a graves problemas respiratorios. Según la historia natural de la enfermedad, la diferencia entre la edad media de diagnóstico (4,7 meses) y la de ingreso en asistencia respiratoria mecánica (5,9 meses) es de 1,2 meses.10 En nuestro caso, la terapia de reemplazo enzimático permitió retrasar el ingreso en asistencia respiratoria mecánica hasta los 16 meses de vida.
La cantidad de alfa-glucosidasa ácida remanente en los pacientes con enfermedad de Pompe (material inmunológicamente reactivo) determina el estado CRIM (sigla en inglés de Cross-reactive immunologic material). Si los pacientes tienen enzima nativa residual, se denominan CRIM-positivos, mientras que si no presentan actividad enzimática alguna se denominan CRIM-negativos. Estos últimos desarrollan altos títulos contra la enzima exógena, transformando la terapia de reemplazo enzimático en menos eficaz. Nuestro paciente no tiene estudiado su estado CRIM, pero la seroconversión durante el primer año de tratamiento, con posterior negativización de los títulos anti-alfa-glucosidasa ácida humana recombinante, supone un estado CRIM positivo, que deberá confirmarse.
La disfunción vesical hallada en el paciente reflejaría el compromiso del músculo liso, situación pocas veces comunicada.
Un apartado especial merecen las consideraciones de los efectos de la terapia de reemplazo enzimático sobre el músculo esquelético. Nuestro paciente pudo rolar, sostener la cabeza y sentarse sin apoyo. No obstante -considerando la eficacia del tratamiento sobre el músculo cardíaco- los resultados obtenidos sobre el músculo esquelético son menores a los esperados. Varios hechos podrían explicar este diferente comportamiento: por un lado, el volumen de músculo esquelético constituye aproximadamente el 40% de la masa corporal. Por otra parte, el músculo cardíaco tiene más receptores de manosa-6-fosfato (median la captación enzimática) que el músculo esquelético.15 Además, podrían existir otras vías de captación enzimática (que involucren diferentes receptores en ambos tipos musculares) o ambos tejidos podrían tener una habilidad diferencial para eliminar el glucógeno acumulado.15
La terapia de reemplazo enzimático es un tratamiento compasivo para una enfermedad grave, que muestra resultados variables en función de cada paciente, por lo que se justifica ofrecer este tratamiento. El aspecto ético es complejo y, ante el agravamiento o complicaciones de la enfermedad, sería un Comité de Ética el espacio donde se debería discutir la continuidad o no del tratamiento.
CONCLUSIÓN
Se presentó un paciente con enfermedad de Pompe en terapia de reemplazo enzimático de 7 años de edad. El tratamiento prolongó la supervivencia, mejoró el compromiso cardíaco y retrasó el ingreso en asistencia respiratoria mecánica. Inicialmente, se apreció mejoría en la función del músculo esquelético y en la adquisición de pautas madurativas, pero estos cambios fueron insuficientes para evitar la progresión de la debilidad muscular.
Agradecimientos
A los Dres. Gustavo Berri y Diego Micheli por las evaluaciones cardiológicas. A las Dras. Carina Vega y Constanza Kingston por el soporte clínico. A las autoridades del Hospital Alemán y a Genzyme de Argentina por su colaboración para el inicio del tratamiento compasivo precoz. A la familia de S.P. por su confianza.
1. Martiniuk F, Chen A, Mack A, Arbanitopoulos E, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998;79(1):69-72. [ Links ]
2. Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, et al. Characterization of pre-and post-treatment pathology after enzyme replacement therapy for pompe disease. Lab Invest 2006;86(12):1208-20. [ Links ]
3. Chien Y-H, Hwu W-L. A review of treatment of Pompe disease in infants. Biologics 2007;1(3):195-201. [ Links ]
4. Di Rocco M, Buzzi D, Tarò M. Glycogen storage disease type II: clinical overview. Acta Myologica 2007; 26:42-4. [ Links ]
5. Kishnani PS, Howell RR. Pompe disease in infants and children. J. Pediatr 2004;144(5 Suppl):S35-43. [ Links ]
6. Kingston CPC, Sabio Paz V, Solana CL. Miocardiopatía hipertrófica neonatal: una forma de presentación clínica de la enfermedad de Pompe. Arch Argent Pediatr 2006;104(5):438- 440. [ Links ]
7. Amartino H, Painceira D, Pomponio RJ, Niizawa G, et al. Two clinical forms of glycogen-storage disease type II in two generations of the same family. Clin Genet 2006;69(2):187-8. [ Links ]
8. Palisano R, Rosenbaum P, Walter S, Russell D, et al. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 1997;39(4):214-23. [ Links ]
9. Van den Hout HM, Hop W, van Diggelen OP, Smeitink JA, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112(2):332-40. [ Links ]
10. Kishnani PS, Hwu WL, Mandel H, Nicolino M, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006;148(5):671-6. [ Links ]
11. Amalfitano A, Bengur AR, Morse RP, Majure JM, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001;3(2):132-8. [ Links ]
12. Levine JC, Kishnani PS, Chen YT, Herlong JR, et al. Cardiac remodeling after enzyme replacement therapy with acid a-glucosidase for infants with Pompe disease. Pediatr Cardiol 2008;29(6):1033-42. [ Links ]
13. Cook AL, Kishnani PS, Carboni MP, Kanter RJ, et al. Ambulatory electrocardiogram analysis in infants treated with recombinant human acid alpha-glucosidase enzyme replacement therapy for Pompe disease. Genet Med 2006;8(5):313-7. [ Links ]
14. Ansong AK, Li JS, Nozik-Grayck E, Ing R, et al. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med 2006;8(5):297-301. [ Links ]
15. Raben N, Jatkar T, Lee A, Lu N, et al. Glycogen stored in skeletal but not in cardiac muscle in acid alphaglucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther 2002;6(5):601-8. [ Links ]

