










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.110 no.4 Buenos Aires jul./ago. 2012
PRESENTACIÓN DE CASOS CLÍNICOS
Duplicación parcial del cromosoma 4 asociada con coloboma ocular bilateral
Partial duplication of chromosome 4 in a patient with bilateral ocular coloboma
Dr. Adrián Colliaa, Dra. Alejandra Antaclea, Dra. María José Velosob y Dra. María Gabriela Obregónc
a. Sanatorio Mater Dei.
b. Centro de Estudios Genéticos.
c. Hospital Italiano de Buenos Aires. Ciudad Autónoma de Buenos Aires, Argentina.
Correspondencia: Dr. Adrián Collia: adrianus38@hotmail.com.
Conflicto de intereses: Ninguno que declarar.
Recibido: 24-9-2011
Aceptado: 28-2-2012
http://dx.doi.org/10.5546/aap.2012.e59
RESUMEN
La trisomía parcial 4q es una enfermedad cromosómica rara causada por la duplicación de una porción (comúnmente la distal) del brazo largo del cromosoma 4. En la mayoría de los casos resulta de una translocación balanceada de uno de los progenitores, siendo menos frecuente la aparición de novo. Los pacientes presentan diversas características clínicas según el tamaño y sitio específico de la región comprometida. Su asociación con patologías oculares ha sido escasamente comunicada. Presentamos el primer caso de un paciente pediátrico de sexo masculino con una duplicación parcial de novo del segmento proximal del brazo largo del cromosoma 4 (4q12-q22) y coloboma bilateral de iris, retina y nervio óptico.
Palabras claves: Duplicación 4q; Coloboma ocular bilateral; Pediatría.
SUMMARY
Partial trisomy 4q is a rare chromosomal disease. It involves duplication of a portion (particularly the distal one) of the long arm of chromosome 4. In most cases results from a balanced translocation on one single progenitor. The "de novo" appearance is less common. Depending on the size and location of duplicated genetic material, patients may have different clinical manifestations. Associated eye pathology has been scarcely informed. We report on a novel case of a male infant with a proximal "de novo" 4q12-q22 duplication and bilateral iris, retinal and optic nerve coloboma.
Key words: 4q duplication; Bilateral ocular coloboma; Pediatrics.
INTRODUCCIÓN
La trisomía parcial 4q es una enfermedad cromosómica rara causada por la duplicación de una porción (comúnmente 4q22-q35) del brazo largo del cromosoma 4. En la mayoría de los casos resulta de una translocación balanceada de uno de los progenitores, siendo menos frecuente la aparición de novo.1-3
Los pacientes presentan diversas características clínicas según el tamaño y sitio específico de la región comprometida, a saber: anomalías faciales, del pie y mano (pulgares); discapacidad auditiva; convulsiones; microcefalia; retraso mental; trastornos en el crecimiento y desarrollo psicomotor; reflujo ureterovesical; hernia inguinal o umbilical; criptorquidia; malformaciones cardíacas y renales. No obstante, su asociación con patologías oculares ha sido escasamente comunicada.4
La región duplicada es muy variable en los distintos casos, pero se han publicado algunas correlaciones genotipo-fenotipo.5 En términos generales, los pacientes con pequeñas duplicaciones parciales 4q11-q13 son sanos y de estatura normal. La única manifestación clínica aparente es un retraso en el desarrollo motor y diferentes grados de dificultad en el aprendizaje. Cuando la duplicación es más grande y compromete a las bandas cromosómicas 4q21 o 4q22, es más probable que los pacientes presenten problemas cardíacos o renales.5-7
Nuestro objetivo es comunicar el primer caso de un paciente pediátrico de sexo masculino con una duplicación parcial de novo del segmento proximal del brazo largo del cromosoma 4 (4q12- q22) y coloboma bilateral de iris, retina y nervio óptico.
CASO CLÍNICO
Segundo hijo de padres sanos, no consanguíneos, de origen europeo y sin antecedentes familiares relevantes. El padre tenía 34 años y la madre 40 al momento de la concepción. Los estudios prenatales (ecografías, urocultivos y análisis de sangre de rutina) fueron normales en todos los casos.
El paciente nació a las 40 semanas de gestación, por cesárea y con un Apgar 9/10. Su peso al nacer fue de 3300 g (percentil 25-50), su longitud de 48 cm (percentil 25-50) y su circunferencia cefálica de 33 cm (percentil 25). El estudio de pesquisa neonatal y la prueba de otoemisiones acústicas fueron normales.
En su primera consulta con el pediatra en nuestra institución (a los 2 meses de vida), se le diagnosticó estrabismo convergente y nistagmus patológico (Figura 1). Le era imposible fijar los ojos durante el amamantamiento. Tenía una mancha blanca en ambos ojos y carecía de reflejo rojo. Por tanto, fue derivado de urgencia al servicio de oftalmología pediátrica.

Figura 1. Paciente con duplicación parcial 4q12-22. En su fenotipo destaca estrabismo convergente, ambas pupilas con aspecto de ojo de cerradura y anomalías faciales leves: pliegues epicánticos bilaterales, telecanto y puente nasal ancho.
Al examen oftalmológico, presentaba anisocoria con coloboma de iris, esotropia, movimientos oculares anómalos y erráticos, y falta de fijación y seguimiento de objetos y luces. La prueba de Bruckner reveló una leucocoria bilateral, por lo que se le realizó fondo de ojo y diagnosticó coloboma bilateral de iris, retina y nervio óptico. La patología fue posteriormente confirmada por ecografía ocular (EO), resonancia magnética de encéfalo (RME) efectuada bajo anestesia general y monitoreo, potencial evocado visual por flash (PEV) y electrorretinograma (ERG).
RESULTADOS
• EO: cavidad vítrea libre de ecos patológicos; marcada irregularidad de la pared posterior a nivel del nervio óptico en ambos globos oculares, compatible con coloboma posterior completo de nervio óptico, más acentuado en el ojo izquierdo; retina aplicada (Figura 2).
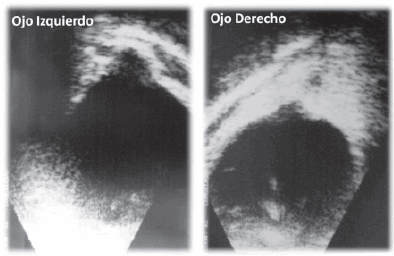
Figura 2. Ecografía ocular (ecógrafo ultrascan de alcon y transductor de 10 Mhz). Se estudiaron todos los meridianos desde la hora 12 y en sentido horario. Luego se realizaron cortes frontales desde pars plana hasta el polo posterior. Se confirmó diagnóstico de coloboma bilateral de nervio óptico y retina.
• RME: la posición de los cristalinos sugiere la existencia de un estrabismo convergente. Los globos oculares mantienen la configuración habitual, con sendos colobomas. No se observan lesiones focales ni cambios difusos de la señal en el parénquima cerebral. El cuerpo calloso y cerebelo mantienen su configuración normal. La mielinización es adecuada para la edad (Figura 3).
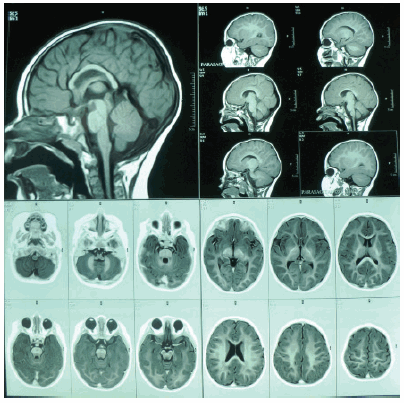
Figura 3. Resonancia magnética de encéfalo. El estudio se efectuó con cortes en los planos de frente, de perfil y transversos. Se utilizaron técnicas para estudiar los tiempos de relajación tisulares T1 y T2. La conclusión fue coloboma bilateral con estrabismo convergente.
• PEV: la forma irregular bilateral y baja amplitud a izquierda sugiere compromiso de la permeabilidad luminosa a izquierda. El ojo derecho presenta una mayor latencia (141 msec) y menor amplitud (11,8 uV) respecto de los valores considerados normales. Lo mismo ocurre con el ojo izquierdo (latencia: 133 msec; amplitud: 3,42 uV).
• ERG: forma irregular y amplitud disminuida bilateral. ERG de tipo subnormal moderado bilateral.
Con todos estos datos se realizó el diagnóstico de disminuido visual. El paciente fue derivado a estimulación visual temprana para mejorar su desarrollo psicomotor y maduración a fin de que pueda aprender a gatear, caminar, tomar objetos y comer solo. También para que en su crecimiento pueda desarrollarse como un niño no vidente aprendiendo posteriormente a manejarse con los nuevos métodos para disminuidos visuales, como es el uso de la computadora con método parlante, y métodos más antiguos como el braille. Por otra parte, cuando su desarrollo motor e intelectual lo permita, se lo enviará a aprender el uso del bastón para que pueda manejarse sólo en la calle y lograr el grado de instrucción posible según su capacidad.
Los padres fueron examinados a fin de identificar la presencia de coloboma u otras malformaciones oculares. Ambos presentaron exámenes oculares normales.
Dados los resultados y la observación de leves anomalías faciales en el niño: pliegues epicánticos bilaterales, telecanto y puente nasal ancho (Figura 1) se decidió realizar un estudio cromosómico. Se obtuvo sangre periférica del paciente y de sus padres a fin de determinar alguna anomalía cromosómica mediante la técnica de bandeo G. El cariotipo de los padres resultó normal. El cariotipo del paciente fue 46,XY,dup(4)(q12q22) de novo (Figura 4) confirmado por hibridización in situ fluorescente (FISH, por su sigla en inglés) con sonda de pintado total del cromosoma 4. Dado el diagnóstico genético se le realizó un electrocardiograma, ecocardiograma, ultrasonido abdominal completo y análisis de sangre y orina, todos informados como normales.
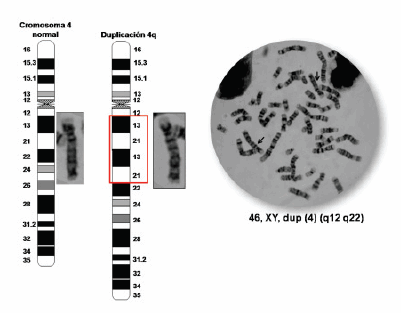
Figura 4. De izquierda a derecha, esquema del cromosoma 4 normal, esquema de la duplicación 4q y foto de los cromosomas en metafase del paciente, confirmando el diagnóstico genético 46,XY,dup(4)(q12q22) de novo.
Se midió la circunferencia cefálica regularmente, la cual permaneció en el percentil 25 durante sus primeros 13 meses de vida.
A la edad de 14 meses, el paciente fue evaluado nuevamente, observándose que presentaba más movimientos oculares anómalos y erráticos, y un crecimiento pondoestatural normal. En cuanto a su desarrollo global, era normal para la edad.
DISCUSIÓN
La duplicación parcial 4q, también conocida como trisomía parcial 4q, implica la existencia de material genético extra del brazo largo del cromosoma 4. La notoriedad y gravedad de las consecuencias dependen de la cantidad de material genético duplicado, de qué parte del brazo cromosómico está comprometido y de qué genes se encuentran alterados por la posición del punto de rotura.
La mayoría de estas duplicaciones resultan de una translocación balanceada de uno de los progenitores. Las duplicaciones de novo, como la de nuestro paciente, cuyos padres son cromosómicamente normales, son poco frecuentes (ocurren durante la formación de las gametas).
En cuanto a las técnicas citogenéticas empleadas, el análisis de array-CGH nos permitiría conocer con más precisión cuál es la región de ADN que se encuentra duplicada en nuestro paciente. Dicha técnica presenta una resolución al menos 10 veces superior a la de un cariotipo convencional, cuya resolución máxima es de 5 megabases. Lamentablemente no es una técnica accesible en nuestro país y, por ello, no ha sido solicitada.
A pesar de las características dismórficas y malformaciones concomitantes presente en muchos pacientes, se cree que no existe un patrón de similitudes tal que permita considerar a las duplicaciones 4q como un síndrome único. Sin embargo, podrían reconocerse distintos síndromes a partir de segmentos cromosómicos cortos, como es el caso de la trisomía parcial 4q31-q35 o "síndrome aurículo-acro-renal".8 Cabe destacar que incluso, dados dos individuos con el mismo genotipo, el fenotipo puede llegar a ser discordante.9
Numerosos autores comunicaron casos de pacientes con pequeñas duplicaciones que comprometen a las bandas 4q31 a 4q33 y están asociadas a efectos fenotípicos leves.10-11 Hegmann y cols., describieron el caso de un niño con una duplicación más larga (4q21.2-q25), que creció normalmente hasta los 3 años, edad a la que notaron un comportamiento impulsivo y breve lapso de atención.12 Elghezal y cols., publicaron acerca de otro niño sano con una duplicación aún más larga (4q25-q34), cuya única manifestación fenotípica fue un leve retraso mental asociado a un déficit del lenguaje.13 En el análisis comparativo realizado por estos últimos autores se sugiere que la región 4q31-q33 podría estar involucrada en el desarrollo de las características dismórficas típicas de esta duplicación, mientras que la banda distal 4q35 podría asociarse con el desarrollo de microcefalia, retraso mental grave y retraso de crecimiento. Shashi y cols., describieron el caso de un paciente con duplicación parcial 4q12-q13 y microcefalia, retraso mental y anomalías faciales leves.5 Carrascosa Romero y cols., presentaron el caso de otro paciente con afectación de la banda distal 4q31-q35 y retraso mental grave-moderado.8 Ergo, estas observaciones no apoyan que la banda 4q35 sea exclusiva en el desarrollo de microcefalia o retraso mental.
Finalmente, vale mencionar que patologías oculares como el estrabismo, nistagmo, oftalmoplejía, morning glory y seudopapiledema se encuentran descriptas en la bibliografía, pero generalmente asociadas a las deleciones 4q.14-15 Nucci y cols., describieron el primer caso de trisomía parcial 4q y morning glory.4 Nuestro paciente es el primer caso comunicado con una duplicación parcial 4q y las manifestaciones oculares antedichas. Asimismo, es el segundo caso comunicado con una duplicación parcial 4q de novo y proximal tan amplia asociado con un fenotipo leve. El primer caso fue descripto por Zollino y cols., en una paciente de 15 años con duplicación parcial 4q13.1- q22.2.6 Como nuestro paciente tiene tan sólo 14 meses de vida, resulta obviamente imposible determinar la ausencia de manifestaciones clínicas a largo plazo, como retraso mental moderado y conducta autodestructiva.6
El examen físico completo de nuestro paciente y la solicitud oportuna de las interconsultas con la oftalmóloga y la genetista permitieron la detección del defecto congénito ocular del niño y su etiología para el asesoramiento familiar, considerando que, al ser una duplicación de novo, el riesgo de recurrencia para próximos hijos de la pareja es bajo.
1. Chen Z, Grebe TA, Guan XY, Notohamiprodjo M, et al. Maternal balanced translocation leading to partial duplication of 4q and partial deletion of 1p in a son: cytogenetic and FISH studies using band-specific painting probes generated by chromosome microdissection. Am J Med Genet 1997;71:160-6. [ Links ]
2. Duval E, van den Enden A, Vanhaesebrouck P, Speleman F. Jumping translocation in a newborn boy with dup(4q) and severe hydrops fetalis. Am J Med Genet 1994;52:214-7. [ Links ]
3. Schrott HG, Sakaguchi S, Francke U, Luzzatti L, et al. Translocation, t(4q-;13q+), in three generations resulting in partial trisomy of the long arm of chromosome 4 in the fourth generation. J Med Genet 1974;11:201-95. [ Links ]
4. Nucci P, Mets MB, Gabianelli EB. Trisomy 4q with morning glory disc anomaly. Ophthalmic Paediatr Genet 1990;11(2):143-5. [ Links ]
5. Shashi V, Berry MN, Santos C, Pettenati MJ. Partial duplication of 4q12q13 leads to a mild phenotype. Am J Med Genet 1999;86(1):51-3. [ Links ]
6. Zollino M, Zampino G, Torrioli G, Pomponi MG. Further contribution to the description of phenotypes associated with partial 4q duplication. Am J Med Genet 1995;57(1):69-73. [ Links ]
7. Mattei M-G, Mattei J-F, Bernard R, Giraud F. Partial trisomy 4 resulting from a complex maternal rearrangement of chromosomes 2, 4 and 18 with interstitial translocation. Hum Genet 1979;51:55-61. [ Links ]
8. Carrascosa Romero MC, García Mialdea O, Vidal Company A, Cabezas Tapia ME, et al. Duplicación parcial del cromosoma 4q (q31, q35): síndrome aurílico-acro-renal. An Pediatr (Barc) 2008;68(4):361-4. Review. [ Links ]
9. Celle L, Lee L, Rintoul N, Savani RC, et al. Duplication of chromosome region 4q28.3-qter in monozygotic twins with discordant phenotypes. Am J Med Genet 2000;94(2):125-40. [ Links ]
10. Goodman BK, Capone GT, Hennessey J, Thomas GH. Familial tandem duplication of bands q31.1 to q32.3 on chromosome 4 with mild phenotypic effect. Am J Med Genet 1997;73(2):119-24. [ Links ]
11. Maltby EL, Barnes IC, Bennett CP. Duplication involving band 4q32 with minimal clinical effect. Am J Med Genet 1999;83(5):431. [ Links ]
12. Hegmann KM, Spikes AS, Orr-Urtreger A, Shaffer LG. Segregation of a paternal insertional translocation results in partial 4q monosomy or 4q trisomy in two siblings. Am J Med Genet 1996;61(1):10-5. [ Links ]
13. Elghezal H, Sendi HS, Monastiri K, Lapierre JM, et al. Large duplication 4q25-q34 with mild clinical effect. Ann Genet 2004;47(4):419-22. [ Links ]
14. Connell P, Brosnahan D, Dunlop A, Reardon W. Bilateral optic disk swelling in the 4q34 deletion syndrome. J AAPOS 2007;11(5):516-8. [ Links ]
15. Parentin F, Fabretto A, Benussi DG, Petix V, et al. Ophthalmic features in a dysmorphic boy with chromosome 4q deletion and duplication. Ophthalmic Genet 2009;30(2):103-5. [ Links ]

