










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.110 no.5 Buenos Aires oct. 2012
PRESENTACIÓN DE CASOS CLÍNICOS
Anemia hemolítica grave causada por hemoglobina Southampton. Presentación de caso clínico
Severe hemolytic anemia due to hemoglobin Southampton. Case report
Dra. Vanesa Avalos Gómeza, Bioq. Silvia Eandi Eberlea, Bioq. Carolina Pepea, Dra. Gabriela Sciuccatia, Bioq. Nerina García Rosolena, Dra. Carolina Cervioa, Dra. Lilian Díaza, Dra. Andrea Candása, Dra. Mariana Bonduela, Lic. Guillermo Piazza a , Lic. Darío Chavesa y Dra. Aurora Feliú Torres
a. Servicio de Hematología-Oncología, Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Buenos Aires.
Correspondencia: Dra. Aurora Feliú Torres: afeliu@garrahan.gov.ar
Conflicto de intereses: Ninguno que declarar.
Recibido: 10-4-2012
Aceptado: 11-5-2012
RESUMEN
Las hemoglobinopatías estructurales son variantes de la hemoglobina caracterizadas por la síntesis de una molécula cualitativamente diferente de la normal. La mayoría son inocuas, mientras que otras ocasionan cambios fisicoquímicos que determinan manifestaciones clínicas de gravedad variable.
En el caso de las hemoglobinas inestables, las alteraciones reducen la solubilidad y facilitan la formación de complejos de hemoglobina desnaturalizada (cuerpos de Heinz) que precipitan, lo cual daña la membrana y destruye prematuramente al eritrocito.
Hasta la actualidad se han descrito 142 hemoglobinas inestables, muchas de ellas ocasionan hemólisis crónica, que puede exacerbarse por infecciones o por la ingesta de medicamentos o drogas oxidantes. La hemoglobina Southampton (también conocida como hemoglobina Casper) es una variante inestable que resulta de la sustitución de un residuo de leucina por uno de prolina, en el codón ß106 (CTG ?CCG], como consecuencia de la mutación c.320 T>C. Presentamos una niña con anemia hemolítica grave, esplenomegalia y requerimiento transfusional debidos a hemoglobina Southampton.
Palabras claves: Anemia hemolítica; Hemoglobina inestable; Hemoglobina anormal.
SUMMARY
Variant hemoglobins are the result of different types of mutations that occur in the globin genes. In many cases, these hemoglobinopathies are harmless, while in others they determine alterations in the physical and chemical properties raising clinical manifestations of variable severity. In the unstable hemoglobinopathies, the changes reduce solubility, inducing the formation of precipitates of denaturated hemoglobin (Heinz bodies), which damage the membrane and finally destroy the red blood cells prematurely. Up to now, more than 142 different unstable hemoglobins have been described, most of them cause chronic hemolysis, increased by infections or drugs. We report the clinical presentation of an unstable hemoglobin (hemoglobin Southampton) in a girl with severe hemolytic anemia, splenomegaly and red blood cell requirement.
Key words: Hemolytic anemia; Unstable hemoglobin; Abnormal hemoglobin.
INTRODUCCIÓN
Las hemoglobinas inestables son el resultado de sustituciones de aminoácidos en sitios críticos de la molécula, que determinan la disminución de su solubilidad y facilitan la formación de complejos de hemoglobina desnaturalizada y precipitada, llamados cuerpos de Heinz.1,2 La inestabilidad de la molécula puede deberse a: 1) sustituciones de aminoácidos en la cavidad del grupo hemo; 2) alteración de la estructura secundaria de la hemoglobina; 3) sustitución de un aminoácido apolar por otro polar, con modificación de la estructura terciaria; 4) alteración en la superficie de contacto alfa/beta que ocasiona la disociación de las cadenas de globina.
Las hemoglobinas inestables tienen un patrón de herencia dominante y dan sintomatología en los heterocigotas; aunque se han descripto, excepcionalmente, algunas formas homocigotas. Se han comunicado mutaciones en diversos grupos étnicos y casi un tercio de ellas son de novo.3
La hemoglobina Southampton es una variante estructural caracterizada por la sustitución de un residuo de leucina por uno de prolina en el codónß106 (CTG ?CCG], que lleva a la disminución de la solubilidad de la proteína. La hemoglobina precipitada y desnaturalizada es responsable tanto del daño de la membrana, como de la disminución de la vida media eritrocitaria.2
Se describen las características clínicas, hematológicas y moleculares en una niña con anemia hemolítica grave causada por esta variante de hemoglobina.
CASO CLÍNICO
Niña de 3 años de edad, que es derivada al Hospital de Pediatría "Prof. Dr. Juan P. Garrahan" con planteo diagnóstico de talasemia mayor, para asesoramiento terapéutico. Primera hija de un matrimonio no consanguíneo, nacida a término por vía vaginal, luego de un embarazo normal. Presentó ictericia durante el período perinatal, que requirió luminoterapia por 7 días, sin aclararse su etiología. A los 12 meses de vida comenzó con ictericia, orinas oscuras y anemia; requirió transfusiones de glóbulos rojos a períodos irregulares. Los síntomas y signos se exacerbaron durante los procesos infecciosos.
Al ingreso, el examen físico mostró palidez e ictericia cutaneomucosa, facies con frente y pómulos prominentes y esplenomegalia. El estudio de la paciente, a tres meses de la última transfusión, y de sus padres, incluyó hemograma (autoanalizador hematológico Sysmex XS800i), examen del frotis de sangre periférica, recuento de reticulocitos, electroforesis de hemoglobina en gel de agar a pH alcalino (SEBIA), cuantificación de hemoglobina A2 por cromatografía de intercambio aniónico (HELENA), cuantificación de hemoglobina fetal por desnaturalización alcalina, metabolismo del hierro, prueba de Brewer (pesquisa de déficit de G6PD), prueba de isopropanol (Carrel y Kay para pesquisa de hemoglobinas inestables) y búsqueda de cuerpos de Heinz.
Los estudios de laboratorio (Tabla 1) revelaron una anemia hemolítica con marcadas alteraciones en la morfología eritrocitaria (Figura 1). En la paciente, tanto la prueba de isopropanol como los cuerpos de Heinz fueron positivos (Figura 2), por lo que se planteó el diagnóstico de anemia hemolítica crónica secundaria a hemoglobina inestable. La evaluación hematológica de los padres fue normal; por lo tanto, y sobre la base de todos estos hallazgos, se descartó el diagnóstico de talasemia mayor.
Tabla 1. Resultados de laboratorio


Figura 1. Frotis de sangre periférica, tinción de May-Günwald-Giemsa, 100X
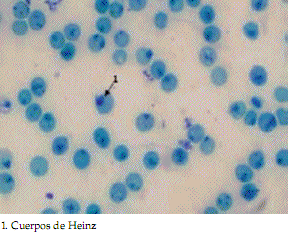
Figura 2. Tinción de azul brillante de Cresilo, 100X
Se procedió a realizar el análisis molecular en ADN total aislado de leucocitos de sangre periférica mediante métodos convencionales. La identificación de mutaciones puntuales en el gen HBB se realizó por PCR y posterior secuenciación con oligonucleótidos cebadores descriptos previamente;4 se detectó la mutación puntual c.320 T>C (Leu107Pro), según nomenclatura sugerida por la Human Genome Variation Society. Esta mutación, hallada en forma heterocigota en la paciente, determina la expresión de la hemoglobina Southampton (ß106(G8)Leu?Pro, CTG?CCG). La mutación no fue detectada en los padres de la paciente, cuya filiación fue confirmada por análisis de 16 microsatélites altamente informativos (AmpFl STR IdentifilerTM PCR Amplification Kit, Applied Biosystems, EE.UU.), lo cual indicó la presencia de una mutación de novo con expresión fenotípica dominante.
El análisis molecular incluyó, además, la búsqueda de alteraciones dentro del cluster alfa, a fin de descartar la posibilidad de co-herencia con la alteración identificada en el gen HBB. Las deleciones más frecuentes [-a3.7; -a4.2; --MED; (-a)20.5] fueron analizadas mediante la técnica GAP-PCR 5 y la identificación de mutaciones puntuales en los genes HBA2 y HBA1 se realizó por PCR-secuenciación.6 Dichos análisis descartaron la presencia de alteraciones en las regiones del cluster alfa analizadas.
DISCUSIÓN
La hemoglobina Southampton, también conocida como hemoglobina Casper, fue descrita por Hyde y cols., en 1972.7 Posteriormente se comunicó en cinco pacientes en todo el mundo, entre ellos, en un niño argentino.8-11
La sustitución de prolina por leucina en la posición G8 106 de la cadena de beta globina interrumpe la secuencia helicoidal y distorsiona gravemente la estructura terciaria de la molécula de hemoglobina, en el punto de unión con el hemo. Se produce así la pérdida del grupo hemo de las subunidades afectadas. Los grupos hemo libres inducen la desnaturalización y posterior precipitación de la molécula de hemoglobina, lo que genera el daño de la membrana y la destrucción temprana de los eritrocitos; cuyo resultado es una anemia hemolítica crónica.3
La hemoglobina Southampton es una variante estructural eléctricamente silente, esto determina la ausencia de banda anómala en la electroforesis de hemoglobinas. La desnaturalización selectiva y posterior remoción de la variante inestable, puede dar como resultado una cuantificación de hemoglobina A2 falsamente aumentada, como se observó en la paciente.2
La prueba de inestabilidad (Carrel y Kay) positiva y la detección de los cuerpos de Heinz pone en evidencia la inestabilidad de la molécula de hemoglobina. Si bien las pruebas pueden ser negativas en pacientes recientemente transfundidos o no esplenectomizados, en la paciente arrojaron datos positivos, lo cual facilitó el diagnóstico hematológico. El análisis molecular define al defecto y posibilita el consejo genético familiar. Como la mayoría de las hemoglobinas inestables, la paciente presenta una mutación de novo, corroborada tanto por el estudio molecular normal de sus padres como por el estudio confirmatorio de la paternidad. Sin embargo, no puede descartarse la presencia de un mosaicismo germinal en alguno de los padres de la niña.
Las manifestaciones clínicas y hematológicas de la hemoglobina Southampton no difieren de las que se observan en otras anemias hemolíticas crónicas, a saber: empeoramiento de la anemia desencadenada por infecciones o por la ingesta de fármacos oxidantes, crisis aplásica secundaria a infección por parvovirus y litiasis vesicular.1-3
Por tratarse de una hemoglobina inestable, además de la suplementación con ácido fólico, los pacientes deben evitar la ingesta de alimentos o de fármacos potencialmente oxidantes. Las transfusiones de glóbulos rojos se indican de acuerdo a la situación clínica del paciente. La esplenectomía sólo se realiza en aquellos con hemólisis grave, a fin de eliminar o reducir el requerimiento transfusional.2
La hemoglobina Southampton debe ser sospechada en pacientes con anemia hemolítica crónica, con prueba de Carrel y Kay positiva y presencia de cuerpos de Heinz, siendo indispensable la realización del estudio molecular para la caracterización final del defecto de la proteína.
1. Sans-Sabrafen J. Hematología clínica. 5ta. ed. Madrid: Elsevier; 2006; Págs. 224-41. [ Links ]
2. Bain B. Haemoglobinopathy diagnosis, 2nd ed. Massachusetts: Blackwell Publishing; 2006; Págs. 215-32. [ Links ]
3. Steinberg M, Forget B, Higgs D, Weatherall D. Unstable hemoglobins. En: Strinberg MH. Disorders hemoglobin. Genetics, pathophysiology, and clinical management. Second Edition. Cambridge: University Press 2009;Págs.593-7. [ Links ]
4. Roldán A, Gutiérrez M, Cygler A, Bolduel M, et al. Molecular characterization of beta-thalassemia genes in an Argentine population. Am J Hematol 1997;54:179-82. [ Links ]
5. Tan AS, Quah TC, Low PS, Chemg SS, et al. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood 2001;98:250-1. [ Links ]
6. Zorai A, Harteveld CL, Bakir A, Van Delft P, et al. Molecular spectrum of alpha-thalassemia in Tunisia: epidemiology and detection at birth. Hemoglobin 2002;26:353-62. [ Links ]
7. Hyde RD, Hall MD, Wiltshire BG, Lehmann H. Haemoglobin Southampton, 106 (G8) Leu leads to pro: an unstable variant producing severe haemolysis. Lancet 1972; 7788:1170-2. [ Links ]
8. Eandi Eberle S, Noguera NI, Sciuccati G, Bonduel M, et al. Hb Southampton [ß106(G8)Leu?Pro, CTG?CCG] in an Argentinean boy. Hemoglobin 2006;30:401-3. [ Links ]
9. Koler RD, Jones RT, Bigley RH, Litt M, et al. Hemoglobin Casper: ß106 (G8) Leu leads to Pro; a contemporary mutation. Am J Med 1973;55(3):549-58. [ Links ]
10. Wajcman H, Gacon G, Labie D, Koler RD, Jones RT, et al. Isolation and functional characterization of hemoglobin Casper: ß106 (G8) Leu replaced by Pro. Biochemistry 1975;14(22):5017-20. [ Links ]
11. Heintz NH, Howard PL. Hemoglobin Southampton (Casper): characterization of the base mutation. Am J Hematol 1989;30(1):1-3. [ Links ]

