










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075
Arch. argent. pediatr. vol.112 no.5 Buenos Aires oct. 2014
http://dx.doi.org/10.5546/aap.2014.e206
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2014.e206
Citrulinemia de presentación atípica: hipo persistente. A propósito de un caso
Dr. Halil Degirmencioglua, Dr. Mehmet Yekta Oncela, Dr. Sadik Yurttutanb, Dr. Sadrettin Ekmena, Dra. Serife Suna Oguza, Dra. Nurdan Urasa y Dr. Ugur Dilmena,b
a. División de Neonatología, Hospital Universitario Maternal Zekai Tahir Burak, Turquía.
b. Departamento de Pediatría, Facultad de Medicina de Sutcu Imam University, Kahramanmaraş, Turquía.
c. Departamento de Pediatría, Facultad de Medicina de Yildirim Beyazit University, Turquía.
Correspondencia: Dr. Halil Degirmencioglu: hdegirmencioglu@gmail.com
Conflicto de intereses: ninguno que declarar.
Recibido: 30-12-2013
Aceptado: 9-4-2014
RESUMEN
Se informa el caso de un neonato que desarrolló encefalopatía en el transcurso de los primeros tres días de vida. Presentaba hipo persistente, que evolucionó a coma profundo 72 horas después de la admisión al hospital. Los parámetros de septicemia y el análisis del líquido cefalorraquídeo (LCR) fueron normales. Tras la evaluación metabòlica, se confirmó la presencia de hiperamoniemia e hipercitrulinemia. El índice de la concentración de LCR/glicina en plasma era normal. Esto no coincidió con nuestro diagnóstico inicial de hiperglicinemia no cetósica, que suele manifestarse con hipo. Se recomienda tener en cuenta la deficiencia de ácido argininosuccínico sintetasa (ASD por su sigla en inglés; citrulinemia) de inicio neonatal en el diagnóstico diferencial de encefalopatía asociada con hipo durante el período neonatal, lo que sugiere una enzimopatía congénita.
Palabras clave: Citrulinemia; Hipo; Neonato.
INTRODUCCIÓN
La citrulinemia es una enzimopatía congénita de herencia autosómica recesiva poco frecuente del metabolismo del ciclo de la urea causada por una deficiencia de ácido argininosuccínico sintetasa, cuya incidencia estimada es de 1/57 000 nacidos vivos.1 Se manifiesta en tres formas clínicas (neonatal, infantil y adulta) y tres tipos según la anomalía enzimática.2 La hiperamoniemia y la hipercitrulinemia graves pueden producir un deterioro neurológico en la citrulinemia.3,4
El hipo suele ser benigno, pero se considera "persistente" si dura entre 48 horas y un mes o "resistente al tratamiento" cuando dura más de un mes, y puede causar complicaciones.5
Hasta donde sabemos, no se han informado casos de encefalopatía asociada con hipo persistente en neonatos afectados por citrulinemia. Informamos un caso de citrulinemia en un recién nacido con dichas manifestaciones clínicas.
Informe de un caso
Los padres de un neonato de sexo masculino realizan una consulta porque el niño, a los tres días de vida, tiene dificultad para alimentarse, vómitos, letargo y marcada hipoactividad. El neonato había sido normal al nacer y se alimentó adecuadamente durante los primeros dos días de vida. Al tercer día de vida, presentó dificultad para alimentarse y se volvió letárgico, con hipo persistente. Nació por cesárea en nuestro hospital a las 39 semanas y 4 días de gestación, de una madre cuadrigesta de 40 años de edad que no tuvo complicaciones en el embarazo. Su peso al nacer fue de 3030 g, con una puntuación de Apgar de 7-9 al primer y quinto minuto, respectivamente. No tenía antecedentes de consanguinidad, y se había registrado una pérdida fetal. El resto de los hermanos eran saludables. El examen físico no reveló características dismórficas ni anomalías. En primer lugar, tenía hipertonía muscular e hiperreflexia en las extremidades superior e inferior derechas, con hipo persistente. Posteriormente, manifestó hipotonía generalizada, dificultad respiratoria y rápidamente desarrolló episodios de apnea, por lo que requirió asistencia respiratoria mecánica. En los siguientes días, tuvo un deterioro neurológico grave con convulsiones que no respondían al tratamiento y coma a las 72 horas de la hospitalización. El análisis del líquido cefalorraquídeo (LCR) fue normal. Los niveles de amoníaco en suero (761 umol/L) y citrulina en plasma y orina (plasma: 2212,4 mol/L) estaban significativamente elevados, y los niveles de ácido orótico en orina fueron altos a las 152 horas de vida. Los niveles de glicina en el LCR y el plasma fueron normales; el índice de concentración de LCR/glicina en plasma fue normal (0,038), lo que descartó nuestro diagnóstico inicial de hiperglicinemia no cetósica, que suele presentarse con hipo. La orina no tenía un olor específico. La ecografía craneal reveló edema cerebral difuso. El estado cortical se evaluó mediante la electroencefalografía de amplitud integrada (aEEG) no invasiva; el trazado fue normal al momento de la hospitalización (Figura 1a), pero mostró un trazado anormal serio a los seis días de vida (Figura 1b). El neonato recibió tratamiento inmediato con benzoato de sodio y solución parenteral de alto valor calórico, dieta hipoproteica e, inicialmente, diálisis peritoneal. Se usó este último recurso porque no disponíamos de hemodiálisis en nuestro hospital. A pesar del tratamiento intensivo, falleció a los nueve días de vida a causa de una insuficiencia cardiorrespiratoria.
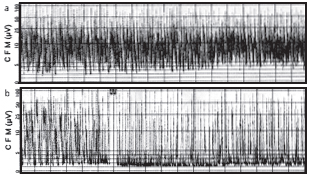
Figura 1. La electroencefalografía de amplitud integrada (aEEG) muestra: (a). Trazado normal al momento de la hospitalización (b). Trazado anormal grave seis horas después de la hospitalización
DISCUSIÓN
El hipo, que es parte de la fase de desarrollo del feto, puede ser un síntoma no específico pocas veces asociado con la irritación del nervio vago y del nervio frénico, trastornos del tronco encefálico, trastornos metabólicos y drogas/ toxinas.6-8
El hipo se produce cuando una inhalación repentina es seguida inmediatamente por el cierre activo de la glotis.6 Al afectar la rama aferente (nervios vago y frénico), la rama central (tronco encefálico, mesencéfalo) y la rama eferente (nervio frénico, glotis y músculos intercostales) del arco reflejo del hipo7 se pueden inducir contracciones involuntarias del diafragma y de los músculos intercostales producidas por el hipo.
Se han sugerido múltiples causas del hipo. La encefalopatía asociada con hipo se considera frecuentemente como señal diagnóstica de la hiperglicinemia no cetósica. A continuación se describe la asociación entre los niveles altos de glicina y el hipo. Se ha demostrado que este efecto enzimático se produce en el cerebro, y la elevación de las concentraciones de glicina produce la acumulación de esta sustancia en el cerebro, que es crítica en la neurotoxicidad.9 El hipo, que probablemente representa un efecto excitador del tronco del encéfalo, podría estar relacionado con el probable efecto excitador de la glicina. Por último, el efecto excitador podría producir convulsiones, hiperexcitabilidad e hipo.9 Habitualmente, se manifiesta en las primeras horas o los primeros días de vida y avanza rápidamente con síntomas neurológicos similares a la citrulinemia.9,10
En principio consideramos que nuestro paciente tenía síntomas compatibles con hiperglicinemia no cetósica al momento de la manifestación de la enfermedad debido a la alta frecuencia de hipo asociado con hiperglicinemia no cetósica. El diagnóstico de laboratorio de hiperglicinemia no cetósica se basa en la determinación simultánea del nivel de glicina en el LCR y el plasma, que demuestra un índice anormal de concentración de LCR/glicina en plasma (por encima de 0,08).10
La citrulinemia es una enzimopatía congénita de herencia autosómica recesiva poco frecuente del metabolismo de la urea causada por la deficiencia de ácido argininosuccínico sintetasa.1,2 El diagnóstico de citrulinemia se basa en el análisis químico de la sangre, el plasma y la orina, que revela niveles elevados de amoníaco, citrulina, glutamina y ácido orótico, como se observó en nuestro paciente.1-4 La realización de un EEG en los lactantes afectados podría mostrar un patrón de salva-supresión típico de la hiperamoniemia neonatal y una correlación cercana de la gravedad del patrón del EEG con el grado de hiperamoniemia y la depresión del nivel de conciencia.3 En forma similar, el aEEG de nuestro paciente mostró actividad de fondo difusamente suprimida (Figura 1b).
La hipercitrulinemia en sí misma posiblemente también sea neurotóxica debido a la inhibición del metabolismo aeróbico de la glucosa.4 Si el trastorno no se reconoce tempranamente ni se trata de inmediato, podrían producirse edema cerebral, aumento de la presión intracraneal y hernia, seguidos de muerte, como en nuestro paciente.1-4 El tronco encefálico no se ve afectado, y la hipercitrulinemia no produce un efecto excitador. La neuropatología de la hipercitrulinemia está asociada con un aumento de la presión intracraneal que se correlaciona con la gravedad de las características neurológicas.1 En un caso de citrulinemia neonatal, se observaron infartos quísticos en la superficie cortical-subcortical, que se consideraron relacionados con la disminución de la perfusión cerebral, secundaria a la presión intracraneal elevada.11
Se demostraron, mediante resonancia magnética, infarto del tronco encefálico y tuberculoma en pacientes adultos con hipo que no respondía al tratamiento.12
Creemos que la causa del hipo en la citrulinemia podría deberse al infarto del tronco encefálico asociado con el aumento de la presión intracraneal. Sin embargo, no fue posible demostrarlo en nuestro paciente con estudios de diagnóstico por imagen debido a su rápido deterioro.
Uno de los puntos débiles de este informe es que nuestro caso se diagnosticó mediante análisis químicos en lugar de análisis de enzimas y de biología molecular. Esto podría tener cierta relevancia ya que existen algunas formas de citrulinemia (asociada con genotipos específicos) que se manifiestan en los primeros años de vida con insuficiencia hepática.
La detección sistemática en los recién nacidos es un programa de salud pública diseñado para detectar, inmediatamente después del nacimiento, enfermedades tratables incluidas en una lista que no son clínicamente evidentes en el período neonatal. Es posible detectar estas enfermedades en la primera etapa de la vida si se amplía la detección sistemática en el lugar donde ocurre el parto.13 La detección sistemática en los recién nacidos solo es útil en los casos en que las manifestaciones clínicas aparecen más tarde. En el caso de las formas clínicas tempranas y graves, como en nuestro paciente, la detección sistemática no es útil.
Hasta donde sabemos, no se han informado casos de encefalopatía asociada con hipo persistente en el período neonatal con la citrulinemia. Es posible que la hipercitrulinemia produzca hipo persistente asociado con encefalopatía. La citrulinemia debería tenerse en cuenta en el diagnóstico diferencial de las enzimopatías congénitas acompañadas de hipo.
1. Wayenberg JL, Vermeylen D, Gerlo E, Pardou A. Increased intracranial pressure in a neonate with citrullinaemia. Eur J Pediatr 1992;151(2):132-3. [ Links ]
2. Sanjurjo P, Rodríguez-Soriano J. Management of neonatal citrullinemia. J Pediatr 1993;123(5):838-9. [ Links ]
3. Clancy RR, Chung HJ. EEG changes during recovery from acute severe neonatal citrullinemia. Electroencephalogr Clin Neurophysiol 1991;78(3):222-7. [ Links ]
4. Bireley WR, Van Hove JL, Gallagher RC, Fenton LZ. Urea cycle disorders: brain MRI and neurological outcome. Pediatr Radiol 2012;42(4):455-62. [ Links ]
5. Smith HS, Busracamwongs A. Management of hiccups in the palliative care population. Am J Hosp Palliat Care 2003;20(2):149-54. [ Links ]
6. Madu AE, Oliver L. Non-ketotic hyperglycinaemia: case report and review of medical literature. J Matern Fetal Neonatal Med 2013;26(5):537-9. [ Links ]
7. Friedman NL. Hiccups: a treatment review. Pharmacotherapy 1996;16(6):986-95. [ Links ]
8. Loft LM, Ward RF. Hiccups: a case presentation and etiologic review. Arch Otolaryngol Head Neck Surg 1992;118(10):1115-9. [ Links ]
9. Tada K, Kure S. Non-ketotic hyperglycinemia: Molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis 1993;16(4):691-703. [ Links ]
10. Hoover-Fong JE, Shah S, Van Hove JL, Applegarth D, et al. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 2004;63(10):1847-53. [ Links ]
11. Martin JJ, Farriaux JP, De Jonghe P. Neuropathology of citrullinemia. Acta Neuropathol 1982;56(4):303-6. [ Links ]
12. al Deeb SM, Sharif H, al Moutaery K, Biary N. Intractable hiccup induced by brainstem lesion. J Neurol Sci 1991;103(2):144-50. [ Links ]
13. Mak CM, Lee HC, Chan AY, Lam CW. Inborn errors of metabolism and expanded newborn screening: review and update. Crit Rev Clin Lab Sci 2013;50(6):142-62. [ Links ]

