










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075
Arch. argent. pediatr. vol.113 no.6 Buenos Aires Dec. 2015
http://dx.doi.org/10.5546/aap.2015.e323
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2015.e323
Hamartomas hipotalámicos: distintas formas de debut. Casos clínicos
Hypothalamyc hamartomas: different ways of clinical debut. Cases report
Dra. Mercedes Cemeli-Canoa, Dra. Marta López Úbedaa, Dr. Antonio de Arriba Muñoza, Dra. Marta Ferrer Lozanoa y Dr. José I. Labarta Aizpúna
a. Unidad de Endocrinología Pediátrica. Hospital Infantil Miguel Servet, Zaragoza, España.
Correspondencia:
Dra. Mercedes Cemeli Cano: merche499@hotmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 29-1-2015
Aceptado: 19-5-2015
RESUMEN
Los hamartomas hipotalámicos son malformaciones no neoplásicas de sustancia gris compuestas por neuronas hiperplásicas. Suelen ser lesiones pequenas localizadas en la base del cerebro, en el piso del tercer ventrículo y, generalmente, asintomáticas. Sin embargo, pueden ocurrir con alteraciones conductuales-cognitivas, crisis epilépticas y/o signos de pubertad precoz central en función de la localización en la que se encuentren.
Se presentan dos pacientes de 2 años 8 meses y 7 años, con presencia de hamartomas hipotalámicos diagnosticados tras el estudio de pubertad precoz central. La paciente de menor edad presenta, además, crisis gelásticas, típicamente asociadas a hamartomas hipotalámicos. Tras los hallazgos clínicos y radiológicos, se trataron con análogos de gonadotropinas, y se observó una regresión de los signos puberales y una no progresión del tamano de los hamartomas.
Palabras clave: Hamartoma hipotalámico; Pubertad precoz central; Epilepsia gelástica.
ABSTRACT
Hypothalamic hamartomas are benign tumors of gray substance composed by hyperplasic neurons. They are usually asymptomatic small masses with extensions into the third ventricular cavity. In some instances they can cause cognitive behavioral alterations, seizures and/or central precocious puberty depending on the location.
Here we present two cases of central precocious puberty due to hypothalamic hamartomas at 28/12 and 7 years of age. The younger patient also presents gelastic seizures, typically associated with hypothalamic hamartomas. After the clinical and radiological findings, they started treatment with GnRH analogues and a regression of the puberty signs without progression in the hamartomas size was observed.
Key words: Hypothalamic hamartomas; Central precocious puberty; Gelastic epilepsy.
INTRODUCCIÓN
El hamartoma hipotalámico (HH) es uno de los hallazgos más comúnmente observados ante la presencia de pubertad precoz central (PPC) en algún momento evolutivo; sin embargo, es poco frecuente como forma de debut.1-4 Su prevalencia en niños y adolescentes se estima en torno a 1-2 casos/100000 habitantes.5 Pueden ser sésiles (intrahipotalámicos) o pedunculados (parahipotalámicos); estos últimos son los que con mayor frecuencia se asocian con PPC. La mayoría son esporádicos, aunque pueden asociarse a otros síndromes congénitos.5,6
El espectro clínico del HH es amplio, desde pacientes asintomáticos hasta presencia de alteraciones endocrinológicas aisladas, como PPC o, incluso, epilepsia gelástica, con pronóstico más desfavorable.5,6
Debido a la presencia de nódulos de pequenas interneuronas gabanérgicas con actividad eléctrica espontánea, el HH presenta actividad epileptógena intrínseca. Las crisis gelásticas son las más características de los HH sésiles en los primeros años de vida y, generalmente, están acompanadas de signos autonómicos.5,6
La PPC producida por HH parece presentarse en edades más tempranas que las producidas por otro tipo de lesiones.1,4 La primera línea de tratamiento serían los agonistas de las gonadotropinas (GnRHa), y se reservarían la radioterapia y la exéresis para casos refractarios al tratamiento farmacológico. El tratamiento quirúrgico ha sido eficaz en un amplio número de pacientes;3 sin embargo, todavía son necesarios estudios a largo plazo que confirmen ese efecto beneficioso mantenido, ya que parece relacionarse con un mayor índice de recidivas y escaso control de los signos de pubertad precoz (PP).
Por ello, nuestro objetivo es presentar dos casos recientes de HH, con motivo de consulta inicial poco frecuente, como es la PP.
CASOS CLÍNICOS
Paciente 1
Nina de 7 años, procedente de Costa de Marfil, remitida para un estudio de PP. Al nacer, tuvo un peso de 4080 g y longitud desconocida. No había antecedentes familiares de PP. Talla genética de 169 cm. Refirieron el inicio de telarquia y pubarquia a los 5 años con menarquia a los 610/12 años. Polifagia, poliuria y polaquiuria, con peor agudeza visual en los últimos meses. Al momento de la exploración física, tuvo un peso de 45,7 kg (+ 4,6 DE) y una talla de 140,5 cm (+ 3,9 DE), con desarrollo puberal adulto (Tanner IV). Menstruaciones regulares. Se realizó un estudio analítico hormonal, con marcadores tumorales negativos (Tabla 1). La valoración oftalmológica con fondo de ojo fue normal. Edad ósea (EO) de 12 años (pronóstico de talla adulta de 155,9 cm). En la resonancia magnética (RM) cerebral (Figura 1), se identificó ocupación de la cisterna supraselar por una masa redondeada dependiente del suelo del tercer ventrículo, de 14 x 13 x 12 mm, compatible con HH del tuber cinereum sin efecto masa.
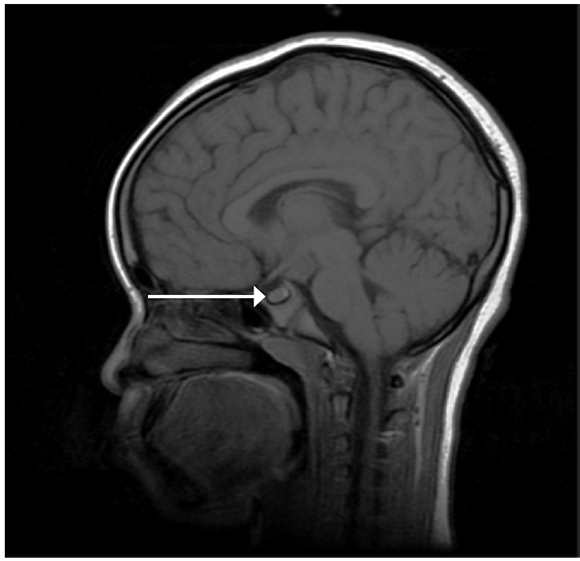
Figura 1. Corte sagital en T1 que muestra hamartomahipotalámico en cisterna supraselar de la paciente 1
Tabla 1. Resumen de las características de los pacientes con hamartomas hipotalámicos de la serie 
Dado el diagnóstico de PPC secundario a HH, se inició un tratamiento con acetato de triptorelina en una dosis inicial de 95,5 µg/kg/28 días intramuscular. Se realizó el seguimiento de la paciente y se observó una regresión importante del estadio puberal, descenso de niveles de LH, FSH y estradiol a valores prepuberales y desaceleración de la velocidad de crecimiento (VC). En el último control, tuvo un peso de 47 kg (+ 3,7 DE) y una talla de 143,4 cm (+ 3,4 DE) con VC de 2,5 cm/año.
En la RM de control, se observó como el HH del tuber cinereum permaneció estable en tamano sin producir complicaciones.
Paciente 2
Nina de 2 años y 8 meses remitida por botón mamario bilateral de rápida progresión en los últimos 3 meses. Sin antecedentes personales ni familiares de interés. Refirieron episodios de irritabilidad ("rabietas") sin causa aparente, que cedían tras 20 minutos, y en ocasiones, alteraciones del sueno. Al momento de la exploración física, presentaba estadio de Tanner III con un diámetro glandular de 2,5 cm sin pigmentación areolar. Flujo vaginal apreciable sin sangrado. Desarrollo psicomotor normal. Peso de 15 kg (+ 0,5 DE) y talla de 97,6 cm (+ 1,2 DE). La EO era de 4 años. Se solicitaron analíticas (Tabla 2), que mostraron activación del eje hipotálamo-hipófisis-gonadal.
Tabla 2. Resumen con los resultados de las analíticas hormonales realizadas al inicio en cada paciente 
En la ecografía ginecológica, se observó un útero y ovarios aumentados de tamano para su edad. En la RM (Figura 2), se apreció una lesión redondeada de 10 mm compatible con HH del tuber cinereum.
Debido a la presencia de "crisis de rabietas", posibles crisis gelásticas, propias también de los HH, se realizó un electroencefalograma, que no mostró grafoelementos patológicos, por lo que se decidió una actitud expectante. Se inició un tratamiento con triptorelina de 98,5 µg/kg/28 días intramuscular a los 210/12 años y se revirtió el estadio puberal a Tanner I, con peso de 19,8 kg (+ 3 DE) y talla de 116 cm (+ 5,8 DE). Los episodios de rabietas mejoraron en número y duración. La RM de control a los 2 años, tras el tratamiento, no mostró variación del HH en senal, morfología ni tamano.
En las Tablas 1 y 2, se representan las características clínicas y analíticas de los dos casos presentados.
DISCUSIÓN
Los HH son malformaciones no neoplásicas de sustancia gris compuestas de neuronas hiperplásicas. Suelen ser lesiones pequenas, de entre 0,5 y 2 cm de diámetro, localizadas en la base del cerebro, en el piso del tercer ventrículo, cerca del tuber cinereum y cuerpos mamilares.
Su frecuencia global es baja, pero su importancia radica en su asociación con epilepsia, problemas cognitivos-conductuales y/o PP (que es una causa inicial rara de ella).5-8
Así como, en mujeres, la causa más frecuente de PP es la idiopática (5% de causa orgánica), en varones, más de un 50% (33%-90%) presenta una causa identificable,7-10 por lo que la realización de una RM es obligada en la mayoría de los casos.
Es más controvertida la indicación de neuroimagen en mujeres; algunos autores la recomiendan en menores de 6 años7,8 o entre 6 y 8 años de forma individualizada. Sin embargo, no se han descrito variables predictoras lo suficientemente seguras como para seleccionar a qué tipo de ninas se les debe realizar.8-10
En nuestro Servicio de Endocrinología Pediátrica, del total de pacientes con PP en 10 años (n= 136), el 13,2% presentan algún hallazgo en la RM (HH en 27,7% de ellos).
Diferenciamos dos tipos de HH, según su clasificación radiológica: pedunculados y sésiles. Los pedunculados (parahipotalámicos) están anexos al suelo del tercer ventrículo o suspendidos desde el hipotálamo inferior, no desplazan el hipotálamo, son de pequeno tamano y asintomáticos, o pueden debutar como PP. Los sésiles (intrahipotalámicos) engloban y desplazan al hipotálamo y la pared del tercer ventrículo, y asocian crisis epilépticas y alteraciones cognitivas-conductuales.11,12
Su debut como PP es frecuente cuando la localización es la de nuestra primera paciente (a nivel del tuber cinereum). Este trastorno endócrino se describe en varias series de pacientes con HH entre 40%-60% a lo largo de la evolución, que inicialmente son asintomáticos.3,5,11,12
PL: punción lumbar; AFP: alfa feto proteína; TSH: hormona tiroestimulante; LH: hormona luteinizante; FSH: homona folículo estimulante; ACTH: hormona estimulante de la corteza adrenal; IGF-1: factor de crecimiento insulínico tipo 1; IGF-BP3: factor de crecimiento insulínico unido a la proteína 3; S-DHEA: sulfato de dehidroepiandrosterona; B-HCG: subunidad beta de la hormona coriónica gonadotrópica.
La fisiopatología del HH como causa de PP no está aclarada, pero se postula un mecanismo activador en la secreción de la hormona luteinizante humana.5,11 Recientes estudios apoyan la mayor influencia de las características anatómicas del HH en la producción de PP que de la expresión de determinadas moléculas, tales como GnRH, TGF a y KISS1.13 Además, el HH no se suele asociar con otras alteraciones endócrinas (déficit de crecimiento, diabetes insípida, hipogonadismo, etc.), a diferencia de otras patologías hipotalámicas.
La primera línea de tratamiento para la PPC asociada a HH es la farmacológica. Los GnRHa han demostrado su efectividad y seguridad,12,14 pero al ser un tratamiento a largo plazo y de elevado costo, se intenta buscar alternativas terapéuticas, como la exéresis. En esta línea, existen varios trabajos, pero con escasos resultados sobre su efectividad a largo plazo. Por ello, se recomienda individualizar el tratamiento, lo que puede indicar la resección inicial en HH < 15 mm y pedunculados fácilmente resecables.14
En los casos descritos, ninguna paciente fue candidata a cirugía como primera opción. En ninguna se constató un cambio en el tamano del HH, tras tratamiento farmacológico, pero sí que hubo una mejoría del estado puberal y de la velocidad de crecimiento. De los pacientes con patología subyacente, dos no recibieron tratamiento con GnRHa por el escaso beneficio que se iba a conseguir en cuanto a su talla diana.
El debut típico de los HH es como crisis epilépticas (gelásticas), tal y como lo hizo la segunda paciente. Se describen como episodios breves, estereotipados de risa o llanto y que pueden acompanarse de signos autonómicos.5,15 Pueden confundirse con sonrisa, cólicos del lactante o trastornos del sueno, como ocurrió en nuestra paciente. Suelen ser refractarias al tratamiento antiepiléptico, incluso en dosis altas o en asociaciones, por ello la ablación del HH ha demostrado un mejor control de las crisis y de los problemas cognitivos-conductuales.5,14 Se ha descrito su evolución a una epilepsia parcial en torno a los 4-10 años,5 de ahí la importancia de su seguimiento.
Como conclusión, se observa una baja frecuencia de HH como causa inicial de PPC y crisis epilépticas. Sin embargo, son entidades para tener en cuenta por su tratamiento dificultoso, fundamentalmente cuando asocian crisis refractarias al tratamiento médico. La primera línea de tratamiento de HH no complicados continúan siendo los GnRHa. La cirugía se reserva para casos refractarios o cuando exista clínica por compresión del HH sobre estructuras adyacentes.
1. Chung EM, Biko MD, Schroeder JW, Cube R, et al. Precocius puberty: radiologic-pathologic correlation. Radiographics 2012;32(7):2071-99. [ Links ]
2. Ng SM, Kumar Y, Cody D, Smith CS, et al. Cranial MRI scans are indicated in all girls with central precocious puberty. Arch Dis Child 2003;88(5):414-8. [ Links ]
3. Rivarola MA, Belgorosky A, Mendilaharzu H, Vidal G. Precocious puberty in children with tumours of the suprasellar and pineal areas: organic central precocious puberty. Acta Paediatr 2001;90(7):751-6. [ Links ]
4. Trivin C, Couto-Silva AC, Sainte-Rose C, Chemaitilly W, et al. Presentation and evolution of organic central precocious puberty according to the type of CNS lesion. Clin Endocrinol (Oxf) 2006;65(2):239-45. [ Links ]
5. Castano De La Mota C, Martín Del Valle F, Pérez Villena A, Calleja Gero ML, et al. Hamartoma hipotalámico en la edad pediátrica: características clínicas, evolución y revisión de la literatura. Neurología 2012;27(5):268-76. [ Links ]
6. Castro LH, Ferreira LK, Teles LR, Jorge CL, et al. Epilepsy syndromes associated with hypothalamic hamartomas. Seizure 2007;16(1):50-8. [ Links ]
7. Soriano-Guillen L, Argente J. Pubertad precoz central: aspectos epidemiológicos, etiológicos y diagnóstico-terapéuticos. An Pediatr (Barc) 2011;74(5):336.e1-336.e13. [ Links ]
8. Soriano-Guillén L, Corripio R, Labarta JI, Canete R, et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J Clin Endocrinol Metab 2010;95(9):4305-13. [ Links ]
9. Stanhope R. Gonadotrophin-dependant precocious puberty and occult intracranial tumors: which girls should have neuro-imaging? J Pediatr 2003;143(4):426-7. [ Links ]
10. Chalumeau M, Hadjiathanasiou CG, Ng SM, Cassio A, et al. Selecting girls with precocious puberty for brain imaging: validation of European evidence-based diagnosis rule. J Pediatr 2003;143(4):445-50. [ Links ]
11. Téllez-Zenteno JF, Serrano-Almeida C, Moien-Afshari F. Gelastic seizures associated with hypotalamic hamartomas: an update in the clinical presentation, diagnosis and treatment. Neuropsychiatr Dis Treat 2008;4(6):1021-31. [ Links ]
12. Striano S, Striano P, Sarappa C, Boccella P. The clinical spectrum and natural history of gelastic epilepsy-hypothalamic hamartoma syndrome. Seizure 2005; 14(4):232-9. [ Links ]
13. Chan YM, Fenoglio-Simeone KA, Paraschos S, Muhammad L, et al. Central precocious puberty due to hypothalamic hamartomas correlates with anatomic features but not with expression of GnRH, TGFalpha, or KISS1. Horm Res Paediatr 2010;73(5):312-9. [ Links ]
14. Li CD, Luo SQ, Gong J, Ma ZY, et al. Surgical treatment of hypothalamic hamartoma causing central precocius puberty: long-term follow-up. J Neurosurg Pediatr 2013;12(2):151-4. [ Links ]
15. García-Morales I, Marinas A, del Barrio A, Álvarez-Linera J, et al. Hamartomas hipotalámicos: características clínicas. Electroencefalograma y resonancia magnética cerebral en 10 pacientes. Neurología 2007;22(1):11-8. [ Links ]

