










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.3 Buenos Aires jun. 2016
http://dx.doi.org/10.5546/aap.2016.e138
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2016.e138
Tos e hipoxemia como manifestación clínica de la proteinosis alveolar. Reporte de caso clínico
Cough and hypoxemia as clinical manifestation of pulmonary alveolar proteinosis. Clinical case report
Dra. Mary Nietoa, Dra. Manuela Dicembrinob, Dr. Rubén Ferraza, Dr. Fernando Romagnolic, Dra. Hilda Giugnob, Dra. Glenda Ernstd, Dra. Monica Siminoviche, Dr. Claudio Castañosb y Dr. Hugo Bottoa
a. Servicio de Endoscopía Respiratoria. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Ciudad Autónoma de Buenos Aires, Argentina.
b. Servicio de Neumonología. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Ciudad Autónoma de Buenos Aires, Argentina.
c. Servicio de Neumonología. Hospital de Niños D. Villegas, Tandil, Buenos Aires, Argentina.
d. Servicio de Neumonología. Hospital Británico, Buenos Aires, Argentina.
e. Servicio de Anatomía Patológica. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Ciudad Autónoma de Buenos Aires, Argentina.
Correspondencia: Dra. Manuela Dicembrino: manu_dice@hotmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 11-8-2015
Aceptado: 12-11-2015
RESUMEN
La proteinosis alveolar es una enfermedad pulmonar crónica poco frecuente, especialmente en pediatría, caracterizada por la acumulación anormal de lipoproteínas y derivados del surfactante en el espacio intraalveolar, que genera una grave reducción del intercambio gaseoso. La forma de presentación idiopática constituye más del 90% de los casos relacionados con un fenómeno de autoinmunidad, con producción de anticuerpos dirigidos contra el receptor del factor estimulante de colonias de granulocitos y macrófagos.
Se presenta un caso clínico de una niña de 4 años de edad tratada por neumonía atípica con evolución desfavorable por hipoxemia persistente. El diagnóstico se obtuvo a través del estudio anatomopatológico de la biopsia pulmonar por toracotomía. Se llevaron a cabo 17 lavados broncopulmonares mediante endoscopía respiratoria y la paciente evidenció franca mejoría clínica.
Palabras clave: Proteinosis alveolar pulmonar; Hipoxemia; GM-CSF.
ABSTRACT
Alveolar proteinosis is a rare chronic lung disease, especially in children, characterized by abnormal accumulation of lipoproteins and derived surfactant in the intra-alveolar space that generates a severe reduction of gas exchange. Idiopathic presentation form constitutes over 90% of cases, a phenomenon associated with production of autoimmune antibodies directed at the receptor for granulocyte-macrophage colony-stimulating factor. A case of a girl of 5 years of age treated because of atypical pneumonia with unfavorable evolution due to persistent hypoxemia is presented. The diagnosis is obtained through pathologic examination of lung biopsy by thoracotomy, as treatment is carried out by 17bronchopulmonary bronchoscopy lavages and the patient evidences marked clinical improvement.
Key words: Pulmonary alveolar proteinosis; Hypoxemia; GM-CSF.
INTRODUCCIÓN
La proteinosis alveolar (PA) es un síndrome infrecuente, en especial, en pediatría, caracterizado por la acumulación anormal de lipoproteínas y derivados del surfactante en el espacio intraalveolar, que genera una grave reducción del intercambio gaseoso.1
Se presenta un caso clínico de una paciente derivada al Hospital "Prof. Dr. Juan P. Garrahan" para el estudio de una neumonía con evolución desfavorable por hipoxemia persistente.
CASO CLÍNICO
Paciente femenino de 4 años y 7 meses de edad, recién nacida a término (RNT; 39 s.), con peso adecuado para la edad gestacional (PAEG; 3370 g). Tuvo mal progreso de peso desde el año, a pesar de una buena actitud alimentaria. Presentó 2 episodios de obstrucción bronquial leves a los 2 años de vida, sin otros antecedentes de relevancia. No tenía antecedentes familiares ni de exposición ambiental.
A los 4 años y 6 meses de edad, se internó por neumonía atípica con hipoxemia, sin aislamiento microbiológico, y fue tratada con oxígeno suplementario por cánula nasal, amoxicilina, claritromicina y oseltamivir en dosis habituales. Luego de 15 días de internación, la paciente evolucionó con persistencia de tos seca, taquipnea e hipoxemia (sat. O2 en aire ambiente -AA-de 89%). La radiografía de tórax evidenció un infiltrado intersticio-alveolar bilateral sin cambios con respecto al ingreso (Figura 1), por lo que se realizó una tomografía de tórax de alta resolución (TAC tórax AR), que mostró un aumento del grosor de los septos interlobulillares, con ocupación alveolar en parches, bilateral en empedrado (Figura 2). Se la derivó al Hospital Garrahan para su estudio.
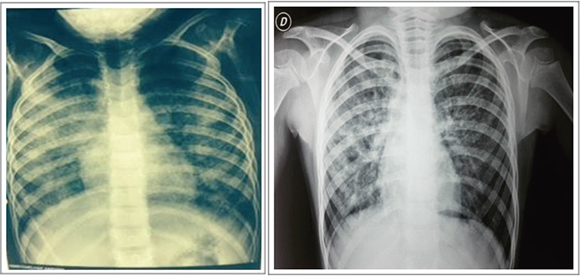
Figura 1. Radiografía de tórax
Compromiso bilateral con infiltrado intersticial fino, difuso y simétrico. Radiografía al momento del diagnóstico (izquierda) y al ano del tratamiento (derecha).
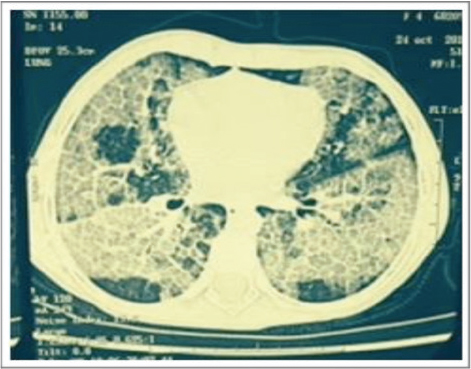
Figura 2. Tomografía de tórax de alta resolución
Compromiso bilateral con infiltrado intersticial fino, difuso y simétrico. Radiografía al momento del diagnóstico (izquierda) y al ano del tratamiento (derecha).
Al ingresar, la paciente se encontraba en buen estado general, delgada (14 kg, Pc 3), con frecuencia respiratoria de 45/minuto, frecuencia cardíaca de 105 latidos por minuto (lpm), sat. O2 AA de 89% (con O2 de 2 L/min: 98%). Tenía buena entrada de aire bilateral sin ruidos agregados. Sin rigidez torácica o hipocratismo digital.
Bajo el diagnóstico de enfermedad pulmonar difusa, se ampliaron los estudios: hemograma, hepatograma, urea y creatinina, dosaje de inmunoglobulinas, análisis de la hormona estimulante de la tiroides (thyroid stimulating hormone; TSH, por sus siglas en inglés), tiroxina y triyodotironina, proteinograma electroforético, C3 y C4, factor reumatoide, anticuerpos antinucleares, antimembrana basal glomerular. También se realizaron derivado proteico purificado (purified protein derivative; PPD, por sus siglas en inglés), cultivo para microbacterias en lavados gástricos y para hongos en secreciones respiratorias, prueba de sudor, dosaje de alfa 1 antitripsina y serologías virales. Todos arrojaron valores normales. La prueba de marcha de 6 minutos y la función pulmonar mediante espirometría, pletismografía y difusión de monóxido de carbono (DLCO) no se realizaron por falta de colaboración de la paciente (probablemente, por su corta edad).
En búsqueda de la etiología, se realizó una endoscopía respiratoria con lavado broncoalveolar (LBA), en la cual se rescató material blanquecino y espeso (Figura 3). Los resultados de los cultivos fueron negativos. El informe de Anatomía Patológica describió material con escasa celularidad, no concluyente. Con sospecha de proteinosis alveolar, por las características macroscópicas, se llevó a cabo la biopsia pulmonar (por toracotomía), que evidenció espacios alveolares distendidos ocupados por material amorfo granular, tinción de ácido periódico Schiff (Periodic Acid-Schiff; PAS, por sus siglas en inglés) positiva, que contribuyeron a la confirmación del diagnóstico presuntivo. Se investigó la presencia de anticuerpos séricos anti factor estimulante de colonias de granulocitos y macrófagos (granulocyte-macrophage colony stimulating factor; GM-CSF, por sus siglas en inglés) mediante un ensayo inmunoenzimático (ELISA), que arrojó un resultado negativo.
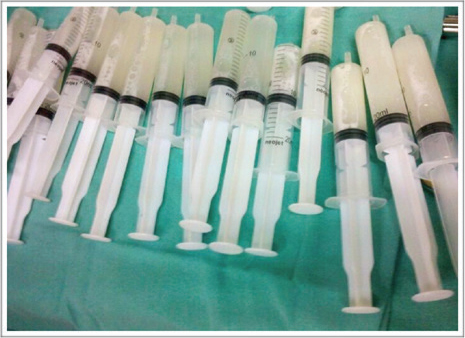
Figura 3. Aspecto macroscópico del material extraído en el lavado broncoalveolar.
Material fluido de aspecto opaco y lechoso.
Como tratamiento, se instauró oxígeno suplementario (0,5 L/min/24 h) y LBA bilaterales, al inicio semanal y luego cada 3 semanas, con un total de 17 procedimientos realizados durante 12 meses. En cada ocasión, se instilaron 500 ml de solución fisiológica a temperatura ambiente, y se rescató más del 90% del material. Se utilizó instrumental flexible, fibrobroncospio Storz (3,5 mm de diámetro externo) a través de una máscara laríngea o tubo orotraqueal. No se observaron complicaciones por los procedimientos efectuados. Este tratamiento se basó en la remoción del contenido alveolar a través del lavado, por lo que se consideró sintomático, sin modificar la fisiopatología de la enfermedad.2
La paciente evolucionó favorablemente, con buen estado general y actitud alimentaria (aumentó 7 kg en un año). En su último control, se evidenciaron valores espirométricos sugestivos de incapacidad ventilatoria restrictiva (IVR) de grado leve: capacidad vital forzada (CVF) de 0,80 (75%), volumen espiratorio forzado en el primer minuto (VEF1) de 0,77 (77%), VEF1/CVF de 96. La prueba de marcha de 6 minutos estuvo dentro de los valores normales (distancia recorrida: 340 m. Sat. O2 mínima: 94%; máxima: 99%). También evidenció menor requerimiento de O2 suplementario (O2: 0,25 L/min durante la marcha y la alimentación; en reposo, la Sat. O2 AA fue de 97%). En la radiografía de tórax, persistieron los infiltrados intersticio alveolares (Figura 1).
La paciente continuó bajo el mismo tratamiento; actualmente, con LBA cada 2 meses, con aclaramiento del material bronquioalveolar rescatado.
PROTEINOSIS ALVEOLAR
Síndrome infrecuente (prevalencia de 0,37 por millón), encuadrado dentro de los desórdenes del catabolismo del surfactante, caracterizado por una alteración en el metabolismo normal de los surfactantes pulmonares, que contribuye a la acumulación de lipoproteínas y derivados del surfactante en el espacio intraalveolar, que altera el intercambio gaseoso a nivel alveolar. Generalmente, afecta a adultos (la edad media de diagnóstico es de 39 años) de sexo masculino (relación 2,6:1).2
El mecanismo fisiopatológico es la anormal activación del receptor del GM-CSF, factor clave en los eventos finales de activación de los macrófagos alveolares, que depuran el material lipídico y son fundamentales en el sistema de vigilancia de la inmunidad innata.3 Puede tener dos presentaciones:4
- Primarias: 1) Genéticas: asociadas a mutaciones en las cadenas alfa o beta del receptor del GM-CSF; déficit de los surfactantes SP-B y SP-C o alteraciones de su estado funcional. 2) Idiopá-ticas: constituyen más del 90% de los casos. Fenómeno de autoinmunidad con producción de anticuerpos dirigidos contra el GM-CSF.5-8
- Secundarias: En pacientes con enfermedades hematológicas, inmunodeficiencias, infecciones crónicas o por exposición a polvos inorgánicos y humos tóxicos.
Se manifiesta con tos seca, disnea progresiva, taquipnea, hipoxemia persistente y mal progreso de peso; estos dos últimos son más frecuentemente en niños, síntomas que se evidencian en nuestra paciente. Más de la mitad de ellos presentan rales crepitantes al momento de la auscultación; el 25%, cianosis y dedos en palillos de tambor. Entre el 10% y el 30% se encuentran asintomáticos al momento del diagnóstico.1,5,9
El diagnóstico se aproxima mediante radiografía de tórax, que evidencia infiltrado intersticial fino, difuso, bilateral y simétrico, que se irradia desde los hilios hacia la periferia. La tomografía axial computada (TAC) de tórax muestra áreas de imágenes radiopacas en vidrio esmerilado, bilateral difuso, con distribución perihiliar, aumento de los septos interlobulillares con patrón típico en empedrado (crazy paving). Los hallazgos clínicos y radiográficos, a menudo, sugieren el diagnóstico. La espirometría sugiere una IVR con disminución de la difusión de monóxido de carbono a través de la membrana alveolocapilar en la prueba de DLCO. En muestra de sangre arterial, se evidencia la presión de oxígeno en sangre arterial (PaO2) disminuida con gradiente A-a aumentado.
Según la disponibilidad, se pueden realizar estudios genéticos para identificar mutaciones para SFTPB, SFTPC, ABCA3 o mutación del gen GM-CSF y/o el dosaje de anticuerpos anti-GM-CSF.1
Si bien la biopsia de pulmón a cielo abierto es el estándar de oro para confirmar el diagnóstico, la endoscopía respiratoria con toma de muestras del LBA permite observar, en el 75% de los casos,2 fluido de aspecto opaco y lechoso, macrófagos alveolares espumosos, grandes cuerpos eosinófilos acelulares y detrito granular positivo para la tinción de Schiff.
Aunque no hay estudios aleatorizados ni prospectivos que evalúen la efectividad de los LBA como tratamiento, se aplican en adultos desde 1960, y se han optimizado las técnicas para llevarlos a cabo. La indicación estaría determinada por la sintomatología del paciente, sea por disnea, que limita la actividad diaria, y/o caída en la oxigenación arterial. Tampoco existe consenso sobre la cantidad y periodicidad de lavados pulmonares para realizar, por lo que es el estado clínico, funcional y radiográfico el que marca la conducta terapéutica.5
La mortalidad se asocia a insuficiencia respiratoria en el 70% de los casos y a sobreinfecciones respiratorias en el 15%-18%, predominantemente en las formas asociadas a la disfunción de GM-CSF, que presentan mayor vulnerabilidad a las bacterias, micobacterias, adenovirus y patógenos oportunistas.10 Algunos autores han reportado una resolución espontánea, que oscila entre 9% y 25% de los casos.2 Los nuevos avances científicos están dirigidos a terapias inmunológicas complementarias al tratamiento, en el caso en que los pacientes presenten la forma autoimnune.1,11
CONCLUSIÓN
Se describe una enfermedad pulmonar infrecuente, la proteinosis alveolar, que presentó como manifestación inicial mal progreso de peso asociado a hipoxemia persistente.
Se expone la importancia de sospechar enfermedades poco habituales ante la evolución tórpida de cuadros clínicos cotidianos.
En gran parte de los casos, la evolución clínica, el patrón imagenológico y el estudio anatomopatológico del LBA son suficientes para arribar al diagnóstico.
Los LBA son el único tratamiento efectuado y han evidenciado una mejoría clínica de la paciente; sin embargo, su frecuencia e interrupción estarán determinadas por su evolución clínica, funcional e imagenológica.
1. Jouneau S, Kerjouan M, Briens E, Lenormand JP, et al. La protéinose alvéolaire pulmonaire. Rev Mal Respir 2014;31(10):975-91. [ Links ]
2. Kattah L, Londono D. Proteinosis alveolar: reporte de caso y revisión de opciones terapéuticas para el 2013. Rev Colom Neum 2013;25(2):110-6. [ Links ]
3. Ben-Dov J, Kishinevski Y, Roznman J, Soliman A, et al. Pulmonary alveolar proteinosis in Israel: ethnic clustering. Isr Med Assoc J 1999;1(2):75-8. [ Links ]
4. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med 2002;166(2):215-35. [ Links ]
5. Young LR, Deustch GH. Childhood interstitial lung disease disorders more prevalent in infancy. En: Wilmott RW, Boat TF, Bush A, Chernick V, et al, eds. Kendig & Chernick's Disorders of the Respiratory Tract in Children. 8th ed. Philadelphia: Elsevier; 2012.Págs.800-9. [ Links ]
6. Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care 2011;56(7):1016-28. [ Links ]
7. Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol 2010;135(2):223-35. [ Links ]
8. Suzuki T. Pulmonary macrophage transplantation therapy of hereditary pulmonary alveolar proteinosis. Keio J Med 2015;64(3):51. [ Links ]
9. Kitamura T, Tanaka N, Watanabe J, Uchida, et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/ macrophage colony-stimulating factor. J Exp Med 1999;190(6):875-80. [ Links ]
10. Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 2009;21(5):514-21. [ Links ]
11. Goldstein LS, Kavuru MS, Curtis-McCarthy P, Christie HA, et al. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest 1998;114(5):1357-62. [ Links ]

