










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.3 Buenos Aires June 2016
http://dx.doi.org/10.5546/aap.2016.e142
.PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2016.e142
Hiperglicinemia no cetósica: mutación novedosa en el gen aminometiltransferasa. A propósito de un caso
Dra. Pinar Gencpinara, Dra. Dilek Çavuşoglub, Dr. Ömer Özbeylerc, Dra. Özge Ö. Kayad, Dra. Figen Baydarf, y Prof. Adj. Dra. Nihal Olgac Dundarb
a. Departamento de Neurología Pediátrica, Hospital Universitario y de Investigación de Tepecik, Esmirna, Turquía.
b. Departamento de Neurología Pediátrica, İzmir Kâtip Çelebi Üniversitesi, Esmirna, Turquía.
c. Departamento de Pediatría, İzmir Kâtip Çelebi Üniversitesi, Esmirna, Turquía.
d. Departamento del Centro de Diagnóstico Genético, Hospital Universitario y de Investigación de Tepecik, Esmirna, Turquía.
Correspondencia: Dra. Nihal Olgac Dundar: nodundar@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 14-8-2015
Aceptado: 9-11-2015
RESUMEN
La hiperglicinemia no cetósica es un raro trastorno metabólico autosómico recesivo hereditario causado por una deficiencia en el sistema enzimático de división de la glicina mitocondrial.
Se desconoce la incidencia general de la hiperglicinemia no cetósica, aunque es mayor en ciertas poblaciones, como las del norte de Finlandia (1/12 000) y de la Columbia Británica (1/63 000). Se sabe que son tres los genes que causan hiper-glicinemia no cetósica: GLDC, AMT y GCSH. Las mutaciones en el gen AMT son responsables del 20% de los casos de hiperglicinemia no cetósica. En este artículo describimos una mutación novedosa del codón de terminación (c.565C>T, p.Q189*) del gen AMT en un niño de cuatro meses de vida con hiperglicinemia no cetósica.
Palabras clave: Convulsiones; Hipo; Mutación en el gen AMT; Neonato; Hipotonía.
INTRODUCCIÓN
La hiperglicinemia no cetósica (HNC) es un error de herencia autosómica recesiva del metabolismo de la glicina que produce la acumulación de grandes cantidades de glicina en los humores corporales y trastornos neurológicos graves inmediatamente después del nacimiento.1 Este trastorno metabólico se debe a un defecto en el complejo de enzimas hepáticas denominado sistema de división de la glicina. Si bien la mutación en el gen GLDC (glicina descarboxilasa) es la más frecuente, en los estudios recientes se ha demostrado que la frecuencia de mutaciones en el gen AMT es similar a la del gen GLDC.2 Presentamos el caso de una mutación novedosa del codón de terminación del gen AMT en la HNC.
Caso
Se hospitalizó a un niño de cuatro meses de edad con hipo, hipotonía y letargo. En sus antecedentes familiares no se informaban abortos espontáneos ni fallecimientos en el período neonatal. En cuanto a los antecedentes personales, se lo había hospitalizado el primer día de vida con convulsiones e hipotonía y luego se inició el tratamiento antibiótico de la sepsis por vía intravenosa. Tenía episodios de apnea por lo que se intubó para recibir asistencia respiratoria. Se inició la administración de fenobarbital y levetiracetam como tratamiento de las convulsiones. Los hemocultivos eran negativos y el valor de proteína C-reactiva era normal.
La electroencefalografía (EEG) mostró actividad de puntas y ondas en las áreas centro-temporales bilaterales. En la resonancia magnética (RM) se observó hemorragia subdural en el área frontal. En el análisis del líquido cefalorraquídeo (LCR) se observó que el recuento de leucocitos y las concentraciones de proteínas y glucosa eran normales. La tinción de Gram, el cultivo y el análisis viral del LCR fueron negativos.
Se realizaron análisis de glucosa, lactato, amoníaco y actividad de la biotinidasa en suero, gasometría, perfil de acilcarnitinas, ácidos orgánicos y cetonas en orina para diagnosticar epilepsia infantil temprana relacionada con trastornos neurometabólicos, por ejemplo, enfermedad de Menkes, síndrome de Zellweger, deficiencia de biotinidasa y aciduria glutárica. La proporción de glicina en LCR/plasma era 0,17 (normal <0,08).
El análisis molecular se realizó mediante secuenciación directa de los genes GLDC, AMT y GCSH. El neonato era homocigoto para una modificación de la secuencia: la sustitución de una citosina por timina en la posición 565 (c.565C>T, p.Q189*) en el gen AMT. Sus padres tenían una mutación heterocigota en el mismo gen (Figura 1).
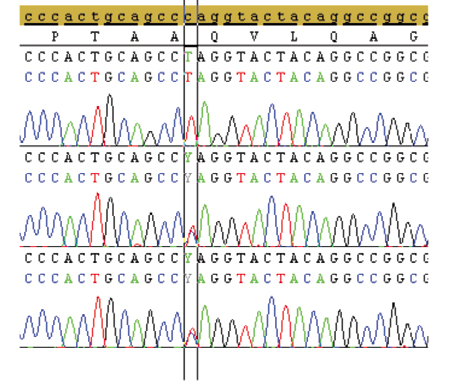
Figura 1. Secuencia de ADN del gen AMT del paciente, la madre y el padre, respectivamente (se marca la región de la mutación).
En las bases de datos, no se mencionaba esta mutación. A los cinco meses de vida, se comenzó la administración de infusión de midazolam debido a las convulsiones resistentes al tratamiento. En ese momento, en el EEG se observaron descargas epilépticas multifocales y encefalopatía epiléptica. Una semana después de haber iniciado la infusión de midazolam, las convulsiones mejoraron y se interrumpió el tratamiento con este fármaco. Al momento del alta, se indicó clonazepam, vigabatrina y fenobarbital. Durante su última hospitalización, cuando tenía 8 meses, ya no tenía convulsiones y recibía topiramato (7,5 mg/kg/día), levetiracetam (34 mg/kg/día) y clobazam (1 mg/kg/día). El paciente tiene hipotonía grave y retraso del desarrollo. A la familia se le comunicaron los resultados de las pruebas durante una sesión de asesoramiento genético. Se estableció que el riesgo de recurrencia del estado homocigoto era del 25% con cada embarazo. Se analizaron opciones de diagnóstico genético preimplantativo/prenatal para los embarazos futuros.
DISCUSIÓN
En este artículo informamos una mutación novedosa (c.565C>T, p.Q189*) en el gen AMT en un recién nacido de sexo masculino. La sustitución de una citosina por timina en la posición 565 produce un codón de terminación y, en consecuencia, se interrumpe la síntesis de la proteína T. La proteína T (enzima que requiere tetrahidrofolato) es un componente del sistema de división de la glicina. Dada la deficiencia de la proteína T, se acumula glicina en el sistema nervioso central, especialmente en el tronco encefálico y la médula espinal, y produce hipotonía y apnea. Por otro lado, el exceso de glicina tiene un efecto agonista sobre el receptor N-metil-D-aspartato (NMDA) excitador del glutamato y explica las convulsiones y los defectos neurológicos a largo plazo. Hasta la fecha, se publicó información sobre 32 pacientes con mutación en el gen AMT (Tabla 1).2-8 Desafortunadamente, debido a que la mayoría de las mutaciones informadas parecen ser raras o privadas, es muy difícil predecir el fenotipo a partir del genotipo. Kure y col.,6 publicaron 11 casos con mutación en el gen AMT. Dos de los 11 pacientes eran homocigotos para las mutaciones, y el resto eran heterocigotos compuestos. Nueve de ellos fueron llevados al hospital durante los primeros días de vida, al igual que en nuestro caso. La mayoría tenía una proporción elevada de glicina en LCR/ plasma. Yılmaz y col.,3 presentaron el caso de un paciente hospitalizado con letargo, convulsiones e insuficiencia respiratoria, con un patrón EEG paroxístico. La mayoría de los casos publicados tenían características similares, como letargo, convulsiones resistentes al tratamiento o patrón EEG paroxístico y proporción elevada de glicina en LCR/plasma (>0,08). De manera interesante, Verissimo y col.,4 publicaron el caso de un paciente con una proporción levemente elevada de glicina en LCR/plasma (0,07) y mutación en el gen AMT. Por lo tanto, es necesario considerar el análisis molecular en los pacientes con sospecha, incluso aunque la proporción de glicina en LCR/ plasma sea normal. Recientemente, Azize y col.,2 publicaron 6 casos con mutación en el gen AMT. Las características clínicas y el pronóstico de estos pacientes eran similares a las de nuestro caso.
Tabla 1. Características clínicas y genéticas de las mutaciones en el gen AMT
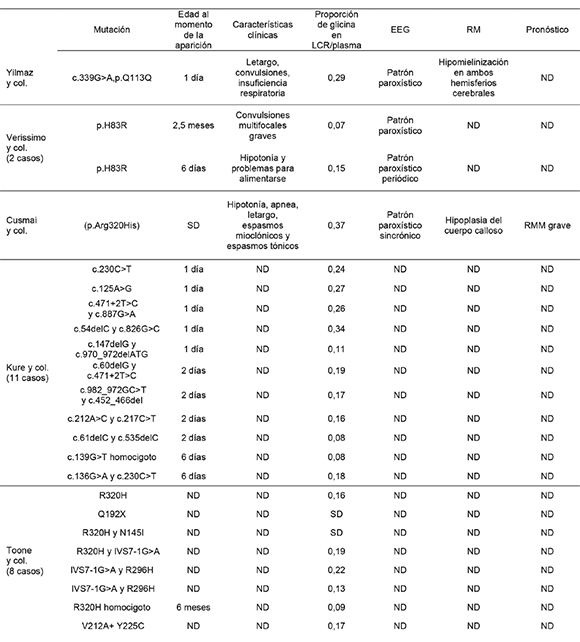
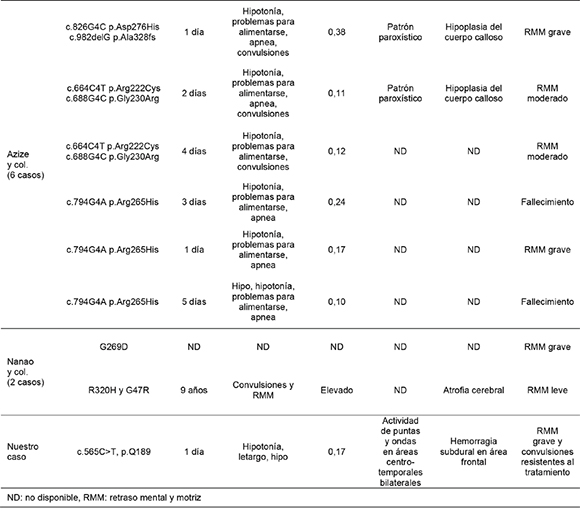
En contraste con estos casos, Nanao y col.,8 publicaron un caso atípico de un paciente heterocigoto compuesto para las mutaciones R320H y G47R en el gen AMT, que tenía un leve retraso mental y motriz.
En la RM de nuestro paciente se observó hemorragia subdural en el área frontal. La base de datos acerca de las RM de pacientes con mutación AMT era insuficiente. Se disponía de datos sobre las características radiológicas solamente de seis pacientes. Tres de ellos presentaban hipoplasia del cuerpo calloso; uno tenía hipomielinización en ambos hemisferios cerebrales; otro, atrofia cerebral, y nuestro
paciente, hemorragia subdural en el área frontal.
No existe una cura para esta enfermedad. En general, el tratamiento incluye benzoato sódico para reducir los niveles plasmáticos de glicina, dextrometorfano para disminuir la excesiva actividad estimulante de la glicina sobre los receptores de NMDA y antiepilépticos para controlar las convulsiones. El desenlace suele ser malo; algunos pacientes fallecen durante el período neonatal, y los que sobreviven suelen tener retraso mental grave y convulsiones resistentes al tratamiento. Recientemente, Swanson y col.,9 publicaron predictores bioquímicos y moleculares para el pronóstico de la hiperglicinemia no cetósica. En este estudio de 124 pacientes, 26 (21%) fallecieron durante el período neonatal o la primera infancia. Los 56 pacientes (45%) con HNC grave tenían un desarrollo muy deficiente uniforme, limitados a sonreír o, raramente, capaces de voltearse de lado a boca abajo. El seguimiento de estos pacientes debe enfocarse en las convulsiones y el neurodesarrollo.
Desafortunadamente, la pesquisa sistemática de HNC en los recién nacidos es probable que no sea beneficiosa ya que no es posible identificar a los neonatos que podrían beneficiarse del tratamiento temprano. Tan y col.,10 demostraron que los neonatos con HNC no suelen tener una concentración de glicina en sangre lo suficientemente elevada a las 48-72 horas de vida como para identificarlos mediante las estrategias de detección sistemática en los recién nacidos.
CONCLUSIÓN
La hiperglicinemia no cetósica es un trastorno metabólico autosómico recesivo muy raro que debe incluirse en el diagnóstico diferencial de los recién nacidos con hipo, convulsiones, espasmos mioclónicos, succión débil e hipotonía. En estos pacientes, debe evaluarse la proporción de glicina en LCR/plasma si presentan manifestaciones clínicas indicativas de alguna forma de hiperglicinemia no cetósica. Después de realizar pruebas metabólicas, debe hacerse un análisis molecular para respaldar el asesoramiento genético y obtener un diagnóstico prenatal en los embarazos posteriores. No fue posible establecer una correlación genotipo-fenotipo sobre la base de los conocimientos actuales. En el futuro, deben realizarse estudios moleculares y funcionales en grandes grupos de pacientes para utilizar los resultados genéticos en la práctica clínica.
1. Hennermann JB. Clinical variability in glycine encephalopathy. Future Neurol 2006;1(5):621-30. [ Links ]
2. Azize NA, Ngah WZ, Othman Z, Md Desa N, et al. Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. J Hum Genet 2014;59(11):593-7. [ Links ]
3. Yılmaz BS, Kor D, Ceylaner S, Mert GG, et al. Two novel missense mutations in nonketotic hyperglycinemia. J Child Neurol 2015;30(6):789-92.
4. Veríssimo C, Garcia P, Simôes M, Robalo C, et al. Nonketotic hyperglycinemia: a cause of encephalopathy in children. J Child Neurol 2013;28(2):251-4. [ Links ]
5. Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, et al. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol 2012; 16(5):509-13. [ Links ]
6. Kure S, Kato K, Dinopoulos A, Gail C, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat 2006;27(4):343-52. [ Links ]
7. Toone JR, Applegarth DA, Levy HL, Coulter-Mackie MB, et al. Molecular genetic and potential biochemical characteristics of patients with T-protein deficiency as a cause of glycine encephalopathy (NKH). Mol Genet Metab 2003;79(4):272-80. [ Links ]
8. Nanao K, Okamura-Ikeda K, Motokawa Y, Danks DM, et al. Identification of the mutations in the T-protein gene causing typical and atypical nonketotic hyperglycinemia. Hum Genet 1994;93(6):655-8. [ Links ]
9. Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol 2015;78(4):606-18. [ Links ]
10. Tan ES, Wiley V, Carpenter K, Wilcken B. Non-ketotic hyperglycinemia is usually not detectable by tandem mass spectrometry newborn screening. Mol Genet Metab 2007;90(4):446-8. [ Links ]

