










Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.3 Buenos Aires jun. 2016
http://dx.doi.org/10.5546/aap.2016.e142
CASE REPORT
http://dx.doi.org/10.5546/aap.2016.eng.e142
Nonketotic hyperglycinemia: novel mutation in the aminomethyl transferase gene. Case report
Pinar Gencpinar, M.D.a, Dilek Çavuºoglu, M.Db, Ömer Özbeyler, M.D.c, Özge Ö. Kaya, M.D.d, Figen Baydan, M.D.a, Nihal Olgac Dundar, Asc. Prof. Dr.b
a. Department of Pediatric Neurology, Tepecik Training of Research Hospital, Izmir, Turkey.
b. Department of Pediatric Neurology, Izmir Katip Çelebi University, Izmir, Turkey.
c. Department of Pediatrics, Izmir Katip Çelebi University, Izmir, Turkey.
d. Department of Genetic Diagnosis Center, Tepecik Training and Research Hospital, Izmir, Turkey.
E-mail address: Nihal Olgac Dundar, M.D. : nodundar@gmail.com
Funding: None.
Conflict of interest: None.
Received: 8-14-2015
Accepted:11-9-2015
ABSTRACT
Nonketotic hyperglycinemia is a rare autosomal recessively inherited metabolic disorder, caused by a deficiency in the mitochondrial glycine cleavage system.
The overall incidence of nonketotic hyperglycinemia is unknown, but is higher in certain populations such as north Finland (1/12,000) and British Colombia (1/63,000). Three genes (GLDC, AMT and GCSH) are known to cause nonketotic hyperglycinemia. Mutations in the AMT gene are responsible for 20% of nonketotic hyperglycinemia cases. We describe a novel stop codon mutation (c.565C>T, p.Q189*) in AMT gene in a four-month male infant with nonketotic hyperglycinemia.
Key words: Seizure; Hiccups; AMT gene mutation; Infant; Hypotonia.
INTRODUCTION
Non-ketotic hyperglycinemia (NKH) is an autosomal recessive inborn error of glycine metabolism, resulting in the accumulation of large amounts of glycine in body fluids and severe neurologic disturbances immediately after birth.1 This metabolic disorder is due to a defect in the liver enzyme complex, termed the glycine cleavage system. Although GLDC (glycine decarboxylase) gene mutation had been known the most seen mutation, recent studies showed that AMT gene mutation is reported with similar frequency to that of GLDC gene.2 We report a novel stop codon mutation in AMT gene in NKH.
CASE
A four-month male infant was hospitalized with hiccups, hypotonia and lethargy. There were no dead of any infant or miscarriage in his family history. In his background, he was hospitalized in his first day with seizure and hypotonia and was commenced on intravenous antibiotics for sepsis. He developed apneic episodes and was then intubated and ventilated. Phenobarbital and levetiracetam was begun for his seizures. Blood cultures were negative and C-reactive protein was normal. Electroencephalography (EEG) indicated spike-wave activity in bilateral centro-temporal areas. Magnetic resonance imaging (MRI) demonstrates subdural hemorrhage in frontal area. Cerebrospinal fluid study (CSF) indicated a normal leukocyte count and protein and glucose levels. The gram stain and culture and viral examination of CSF were negative. Serum glucose, lactate, ammonia and biotinidase activity; blood gas; acylcarnitine profile; urinary organic acids and ketones were performed to diagnose early childhood epilepsy neurometabolic disorders-related such as Menkes disease, Zellweger syndrome, biotinidase deficiency and glutaric aciduria. Cerebrospinal fluid/plasma glycine ratio was 0.17 (normal < 0.08). Molecular analysis was performed by direct sequencing of GLDC, AMT, and GCSH genes. The child was homozygous for a sequence modification: the substitution of a cytosine by thymine at position 565 (c.565C>T, p.Q189*) in the AMT gene. His parents had heterozygous mutation in same gene (Figure 1). This mutation was not mentioned in the databases. He received midazolam infusion because of his intractable seizures, when he was five month old. Multifocal epileptic discharges and epileptic encephalopathy was determined on his EEG at this time. One week after starting midazolam infusion his seizures reduced and midazolam therapy was stopped. He was discharged with clonazepam, vigabatrin and phenobarbital. On his last admission, when he was 8 month old, he was seizure-free with topiramate (7.5 mg/kg/day), levetiracetam (34 mg/kg/day) and clobazam (1 mg/kg/day). He has severe hypotonia and developmental delay. The family was informed about the test results in a genetic counseling session. Recurrence risk for homozygous status was given as 25 percent for each pregnancy. Preimplantation/ prenatal genetic diagnosis options were discussed for further pregnancies.
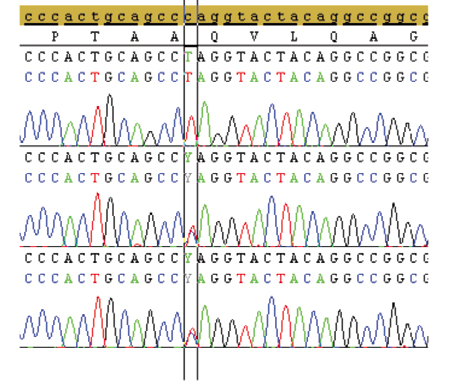
Figure 1. DNA sequence of AMT gene from patient, mother and father respectively (mutation region were marked).
DISCUSSION
Here we report a novel mutation (c.565C>T, p.Q189*) of AMT gene in a male infant. The substitution of a cytosine by thymine at position 565 leads to a stop codon and therefore the synthesis of T protein stops. T protein (a tetrahydrofolate-requiring enzyme) was a component of the glycine cleavage system. Because of T protein deficiency, glycine accumulates in central nervous system, predominantly in the brain stem and spinal cord and causes hypotonia and apnea. On the other hand, excess glycine demonstrates an agonistic effect to glutamate of the excitatory N-methyl-D-aspartate receptor (NMDAR), account for seizures and long-term neurologic defects. Thirty-two patients, who had mutation in AMT gene, were published in the literature up to date (Table 1).2-8 Unfortunately, because most reported mutations seem to be rare or private, it is very difficult to predict phenotype from genotype. Kure et al6 published 11 cases with AMT mutation. Two of 11 had homozygous and the rest had compound heterozygous mutations. Nine of them were presented to hospital in the first few days similarly our case. Most of cases had elevated CSF/Plasma Glycine Ratio. Yılmaz et al3 presented one case, which was admitted with lethargy, seizures and respiratory insufficiency and had burst-suppression pattern on his EEG. Most of published cases had similar clinical features such as lethargy, intractable seizures or burst suppression pattern on the EEG and elevated CSF/Plasma Glycine ratio (>0.08). Interestingly Verissimo et al4 published one case, which had mildly elevated CSF/Plasma Glycine ratio (0.07) and mutation in AMT gene. Therefore molecular analyze should keep in mind in suspected patients even CSF/Plasma Glycine ratio was normal. Recently Azize et al2 published 6 cases with AMT mutation. Clinical features and prognosis of these cases were similar to our case. As distinct from these cases Nanao et al8 published an atypical case with compound heterozygous R320H and G47R mutation in AMT gene, which had mild motor-mental retardation. Our case had subdural hemorrhage in frontal area in his MRI. The database about MRI in AMT mutation was insufficient. There was no available data about radiological features except six patients. Three of them had hypoplasia of corpus callosum, one had hypomyelination in both cerebral hemispheres, one had cerebral atrophy and our case had subdural hemorrhage in frontal area.
Table 1. Clinical and genetic features of AMT gen mutations

There wasn't any curative treatment in this disease. Treatment commonly involves sodium benzoate given to lower plasma glycine levels, dextromethorphan with the intent to reduce the excessive stimulating activity of glycine on N-methyl-D-aspartate receptors and antiepileptic drugs to control seizures. The outcome is poor, some patients dying during the newborn period; survivors usually exhibit severe mental retardation and intractable seizures. Swanson et al9 published biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia recently. In this study of 124 patients, there were 26 patients (21%) who died in the neonatal or early infantile period. The fifty-six patients (45%) with severe outcome NKH represent a uniform very poor development limited to smiling, or rarely able to roll from side to prone. These patients should be follow-up in terms of seizures and neurodevelopmental status. Unfortunately, newborn screening for NKH is unlikely to be beneficial, as those babies who might benefit from early treatment could not be identified. Tan et al10 showed that babies with NKH do not usually have a sufficiently elevated blood glycine level at 48-72 h of life for identification by current newborn screening strategies.
CONCLUSION
Nonketotic hyperglycinemia is a very rare autosomal recessive metabolic condition and should be included as a differential diagnosis in an unwell neonate presenting with hiccup, seizures, myoclonic jerks, poor sucking and hypotonia. Cerebrospinal fluid/ plasma glycine ratio and plasma glycine levels should be evaluated in patients, whose clinical manifestations are suspicious for any form of nonketotic hyperglycinemia. After metabolic testing, molecular analysis should be performed to support genetic counseling and provide prenatal diagnosis for next pregnancies. Genotype-phenotype correlation wasn't able to determine with current knowledge. Further molecular and functional studies in large patient groups are necessary to use genetic results in clinical practice.
1. Hennermann JB. Clinical variability in glycine encephalopathy. Future Neurol 2006;1(5):621-30. [ Links ]
2. Azize NA, Ngah WZ, Othman Z, Md Desa N, et al. Mutation analysis of glycine decarboxylase, aminomethyltransferase and glycine cleavage system protein-H genes in 13 unrelated families with glycine encephalopathy. J Hum Genet 2014;59(11):593-7. [ Links ]
3. Yılmaz BS, Kor D, Ceylaner S, Mert GG, et al. Two novel missense mutations in nonketotic hyperglycinemia. J Child Neurol 2015;30(6):789-92.
4. Veríssimo C, Garcia P, Simôes M, Robalo C, et al. Nonketotic hyperglycinemia: a cause of encephalopathy in children. J Child Neurol 2013;28(2):251-4. [ Links ]
5. Cusmai R, Martinelli D, Moavero R, Dionisi Vici C, et al. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol 2012; 16(5):509-13. [ Links ]
6. Kure S, Kato K, Dinopoulos A, Gail C, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat 2006;27(4):343-52. [ Links ]
7. Toone JR, Applegarth DA, Levy HL, Coulter-Mackie MB, et al. Molecular genetic and potential biochemical characteristics of patients with T-protein deficiency as a cause of glycine encephalopathy (NKH). Mol Genet Metab 2003;79(4):272-80. [ Links ]
8. Nanao K, Okamura-Ikeda K, Motokawa Y, Danks DM, et al. Identification of the mutations in the T-protein gene causing typical and atypical nonketotic hyperglycinemia. Hum Genet 1994;93(6):655-8. [ Links ]
9. Swanson MA, Coughlin CR Jr, Scharer GH, Szerlong HJ, et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol 2015;78(4):606-18. [ Links ]
10. Tan ES, Wiley V, Carpenter K, Wilcken B. Non-ketotic hyperglycinemia is usually not detectable by tandem mass spectrometry newborn screening. Mol Genet Metab 2007;90(4):446-8. [ Links ]

