










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.6 Buenos Aires Dec. 2016
http://dx.doi.org/10.5546/aap.2016.e403
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2016.e403
Niño con síndrome tricorrinofalángico tipo II acompañado de baja estatura
Dra. Filiz Hazana, Dr. Hüseyin A. Korkmazb, Dra. Kanay Yararbaşc, Dr. Wim Wuytsd y Dra. Ajlan Tüküne
Colaborador: Dr. Ali Dülgeroğluf
a. Hospital de Formación e Investigación de Enfermedades y Cirugías Pediátricas Dr. Behçet Uz, Departamento de Genética, Esmirna, Turquía.
b. Hospital de Formación e Investigación de Enfermedades y Cirugías Pediátricas Dr. Behçet Uz, Departamento de Endocrinología Pediátrica, Esmirna, Turquía.
c. Duzen Laboratories Group, Departamento de Genética, Ankara, Turquía.
d. Departamento de Genética Médica, Hospital Universitario de Amberes, Bélgica.
e. Escuela de Medicina de Ankara Üniversitesi, Departamento de Genética Médica, Ankara, Turquía.
f. Hospital de Formación e Investigación ízmir Atatürk, Departamento de Ortopedia y Traumatología, Esmirna, Turquía.
Correspondencia: Dr. Hüseyin A. Korkmaz: drkorkmazanil@hotmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 27-11-2015
Aceptado: 19-5-2016
RESUMEN
El síndrome tricorrinofalángico (STRF) tipo II (sinónimo: síndrome de Langer-Giedion) es un síndrome autosómico dominante raro que afecta genes adyacentes y que se produce como resultado de una microdeleción que abarca los genes EXTl y TRPSl en la banda 8q24 (OMIM 150230). En este síndrome se combinan características de dos trastornos autosómicos dominantes: el síndrome tricorrinofalángico tipo I (OMIM 190350) y la osteocondromatosis múltiple hereditaria tipo I (OMIM 133700). El STRF tipo II se caracteriza por escaso cabello, nariz prominente y de extremo bulboso, surco nasolabial plano y alargado, epífisis de las falanges en forma de cono, retraso de la edad ósea durante la infancia y osteocondromas cartilaginosos múltiples. En este artículo presentamos el caso de un paciente de Turquía con las características clínicas y los signos óseos del STRF tipo II en el que se detectó una deleción de 13,8 Mb en las bandas 8q23.1-8q24.13.
Palabras clave: Síndrome tricorrinofalángico tipo II; Deleción cromosómica; Baja estatura.
INTRODUCCIÓN
Los síndromes tricorrinofalángicos (STRF) son trastornos autosómicos dominantes que se clasifican en tres subtipos (tipo I, II y III) según el patrón de mutación en el gen TRPSl1,2
Las características comunes a los tres tipos son los rasgos faciales únicos, entre otros, cabello escaso y de crecimiento lento, cejas tupidas en el medio y finas en los extremos, nariz de extremo bulboso, surco nasolabial plano y alargado, labio superior delgado con borde de color bermellón y orejas prominentes.2,3 Además, se describieron anomalías óseas en el STRF, entre otras, falanges y metacarpianos cortos (braquidactilia de leve a grave), epífisis en forma de cono, displasia de cadera y baja estatura.2,3 El gen TRPSl, que se encuentra en la banda cromosómica 8q24.1, codifica un miembro atípico de la familia de factores de transcripción GATA y representa un gen candidato para la regulación de la homeostasis ósea.3 El STRF tipo I se produce por una mutación heterocigota o una deleción en el gen TRPSl, mientras que el STRF tipo III es causado por mutaciones heterocigotas de sentido erróneo en este gen. El STRF tipo II, también conocido como síndrome de Langer-Giedion (OMIM 150230), se define como un trastorno de genes adyacentes producto de la pérdida de copias funcionales, no solamente en el gen TRPSl sino también en el gen contiguo EXTl.4 El gen EXTl codifica la proteína transmembranaria tipo 2 ubicada en el aparato de Golgi con actividad glucosiltransferasa.
El gen EXTl parece tener un efecto regulador sobre el crecimiento óseo longitudinal.4
En este artículo presentamos el caso de un paciente con un fenotipo que superponía el STRF tipo II con una deleción cromosómica en las bandas 8q23.1-8q24.13. Ambos padres otorgaron el consentimiento informado por escrito para la publicación de este informe.
Caso clínico
Nuestro paciente era un niño de 12 años derivado a nuestra institución debido a baja estatura, rasgos faciales dismórficos y anomalías óseas. Nació con un peso de 3200 g tras un embarazo a término sin complicaciones. Ambos padres eran turcos, no consanguíneos, y el paciente tenía una hermana sana. No se hallaron casos de anomalías óseas en sus antecedentes familiares. Tenía pie calcáneo valgo bilateral, dislocación bilateral de cadera y hernia inguinal unilateral, que se corrigieron mediante cirugía durante el primer año de vida. A los dos años comenzó a desarrollar osteocondromas, por lo que se lo sometió a intervenciones ortopédicas múltiples. Presentaba retraso en los hitos del desarrollo: logró sentarse al año, pararse a los dos años y caminar a los dos años y medio. Tenía retraso mental moderado.
En el examen físico se observó: peso de 30 kg (percentilo 3-10, puntuación de desviación estándar -1,70), estatura de 120 cm (percentilo <3, puntuación de desviación estándar -4,12) y perímetro cefálico de 45,6 cm (percentilo 1025). Entre sus rasgos faciales se destacaban el nacimiento del cabello en pico de viuda, cabello fino y escaso, cejas tupidas, orejas de implantación baja, nariz bulbosa con columela ancha, surco nasolabial alargado y orejas grandes y de prominencia lateral (Figura 1). Otras características incluían tórax hundido, articulaciones hipermóviles, rodilla valga, osteocondromas múltiples y exceso de piel (Figura 1).
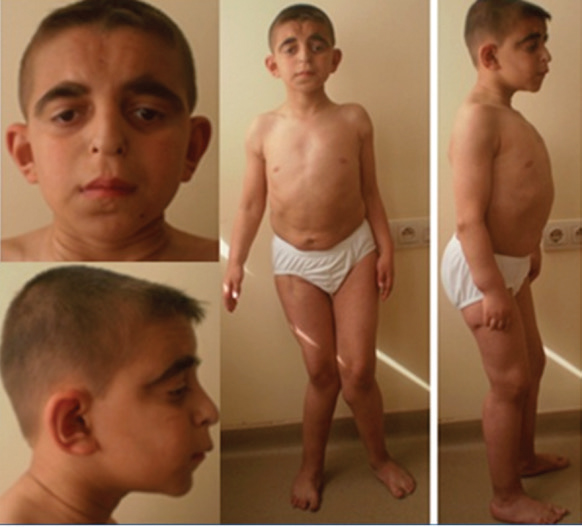
Figura 1: Paciente de 12 años en el que se observan rasgos faciales dismórficos y anomalías óseas
Los resultados de los siguientes análisis de laboratorio fueron normales: hemograma completo, electrolitos, enzimas hepáticas, creatina-fosfoquinasa, calcio, fosfato, fosfatasa alcalina, urea, creatinina, hormonas tiroideas, hormona paratiroidea, 25-hidroxi vitamina D y cromatografía de aminoácidos en sangre y orina. No se observaron anomalías en la ecografía abdominal ni en la resonancia magnética de la cabeza. En la radiografía del esqueleto se observaron osteocondromas múltiples (en las articulaciones de las muñecas, las rodillas y los tobillos), esclerosis en las cabezas femorales, cuellos femorales cortos, irregularidad cortical femoral e irregularidades en la articulación de la cadera (más grave en la cadera derecha). En las radiografías de las manos se observaron retraso marcado de la edad ósea, epífisis en forma de cono en las articulaciones interfalángicas proximales y falanges cortas (Figura 2). El examen del fondo de ojo y la audiometría fueron normales.
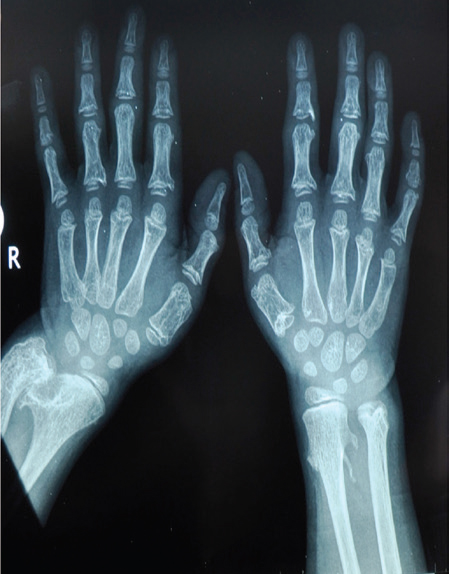
Figura 2: Radiografía de manos y muñecas
Se realizó un análisis citogenético (bandeo cromosómico de Giemsa 550) en un cultivo de linfocitos de sangre periférica conforme a las técnicas citogenéticas estándar. Se detectó una deleción intersticial en el brazo largo del cromosoma 8 que abarcaba las bandas q23 y q24 (Figura 3A). También se confirmó una deleción en el gen EXT1 en la banda 8q24 mediante un análisis de amplificación de sondas dependiente de ligandos múltiples (MLPA, por sus siglas en inglés). Además se realizó un análisis de hibridación genómica comparativa (CGH, por sus siglas en inglés) en una plataforma NimbleGen MS200, que permitió obtener una resolución de detección de una variación en el número de copias (CNV, por sus siglas en inglés) de ≥ 4,7 kb. Mediante la técnica utilizada se logró un mapeo fino de los valores críticos de la deleción y se confirmó la existencia de la deleción entre las bandas 8q23.1 y 8q24.13 (Figura 3B). La deleción se produjo entre las posiciones genómicas 110,332,662-124,170,948. El resultado del análisis citogenético de los padres fue normal, lo que indica que se trataba de una deleción de novo.
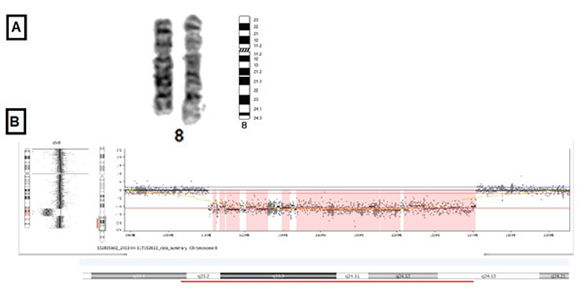
Figura 3: Deleción intersticial en el brazo largo del cromosoma 8 que abarca las bandas q23 y q24
DISCUSIÓN
Este paciente tenía una deleción en las bandas cromosómicas 8q23.1-q24.1 que abarcaba los genes TRPS1 y EXT1, que representan el fenotipo. El gen TRPS1 (OMIM 604386), mapeado en el cromosoma 8q23.1-q24.1, está asociado con el desarrollo y la diferenciación de los huesos, los riñones y los folículos pilosos.1,2,5 El STRF tipo I y tipo III se produce a partir de mutaciones o haploinsuficiencia del gen TRPS1.1-3,6 El STRF tipo I (OMIM 190350) se caracteriza por cabello escaso, nariz de extremo bulboso, surco nasolabial alargado, orejas prominentes, epífisis en forma de cono en las falanges, malformaciones de cadera y baja estatura.6 El STRF tipo II es un síndrome que afecta genes adyacentes, con deleción de los genes TRPS1 y EXT1; se diferencia del STRF tipo I por la presencia de exostosis múltiples y retraso mental.7 El STRF tipo III (OMIM 190351) es similar al tipo I, pero el acortamiento de todas las falanges y los metacarpianos es más grave, la baja estatura y la braquidactilia son graves y no se observan exostosis.3 La mayoría de los casos de STRF tipo II son esporádicos, aunque se observan unos pocos casos hereditarios.3
Las manifestaciones óseas del STRF tipo II incluyen epífisis en forma de cono en las falanges de las manos, que producen deformidad en los dedos, retraso de la edad ósea, osteocondromas y malformaciones de cadera.3 Verheij y col. informaron sobre dos pacientes con una deleción en el brazo largo del cromosoma 8.8 En uno de los casos, la región de la deleción (q24.1-24.3) incluía los genes TRPS1 y EXT1. El paciente presentaba los rasgos faciales del STRF tipo II, pero también anomalías en las manos, incluida la fusión del tercer y cuarto metatarsiano y oligodactilia, coloboma unilateral del iris derecho, estenosis pulmonar y comunicación interventricular. Se sugirió que otros genes presentes en la región eliminada podrían ser la causa de estas características adicionales.8
Nuestro paciente tenía los rasgos faciales y los signos óseos típicos del STRF tipo II. Las manifestaciones clínicas están estrechamente asociadas con el STRF tipo II, el síndrome de deleción de la banda 8q24.1. El paciente tenía una deleción de 13,8 Mb entre las bandas 8q23.1 y 8q24.13.9,10 Uno de ellos, el gen sobreexpresado de nefroblastoma (NOV), se ubica en la banda 8q24.1. La proteína NOV es miembro de la familia CCN (proteínas secretadas ricas en cisteína). Los miembros de la familia CCN se expresan en la matriz extracelular, y tienen un rol crucial en la diferenciación y la función celular, el desarrollo cardiovascular y esquelético, la reparación de lesiones, las fibrosis y el cáncer. Se conoce parcialmente su expresión y función en el tejido esquelético.11 En un estudio realizado en ratones con y sin mutaciones Novdel3 se reveló que los ratones Novdel3+/- y Novdel3-¡- presentaban defectos múltiples en la esqueletogénesis y la formación de articulaciones. Entre las anomalías óseas se observaron fusión de los tarsianos, aplanamiento de la rótula, malformación de la muñeca, dislocación de la cadera y acoplamiento anómalo de las articulaciones, lo que produce laxitud de las extremidades. Los autores sugirieron que un análisis molecular detallado de la diferenciación condrogénica de los embriones Novdel3-/- permitirá esclarecer la función del gen NOV en esta vía.12 Nuestra hipótesis es que la deleción del gen NOV podría tener una función en las anomalías óseas de los seres humanos.
A nivel radiológico, la manifestación más frecuentemente observada en el STRF son las epífisis en forma de cono.1 Las falanges medias en forma de cono tipo 12 son características del síndrome de Giedion, con afinamiento de la periferia hacia la base.13 El signo de excavación está casi siempre presente en los conos tipo 12, con excavación progresiva de la base del cono. Nuestro paciente tenía un cono tipo 12A, que se observa ocasionalmente en el STRF tipo I y III, y casi siempre en el tipo II.13 Sin embargo, en las manos de los pacientes con STRF se observan pulgar trifalángico, que puede estar asociado a polidactilia, acortamiento de los metacarpianos (especialmente el anular y el meñique) y acortamiento de las falanges.14 En nuestro paciente se observaron conos tipo 12A y acortamiento de las falanges. El mecanismo subyacente de la asociación entre el STRF tipo II y las epífisis en forma de cono y los pulgares bífidos y cortos es el gen TRPSI, que es responsable de regular la homeostasis ósea. Estudios futuros podrían ayudarnos a comprender la función del gen TRPSI en la heterogeneidad de los signos óseos.
A modo de conclusión, este breve informe sobre un paciente turco con STRF tipo II es relevante debido a la confirmación de la gran deleción en las bandas 8q23 y q24 mediante un análisis de matriz de CGH. La amplia variación en el tamaño de las deleciones y en los demás genes en las regiones eliminadas podría ser la causa de las características adicionales de los pacientes con STRF tipo II.
1. Lûdecke HJ, Wagner MJ, Nardmann J, La Pillo B, et al. Molecular dissection of a contiguous gene syndrome: localization of the genes involved in the Langer-Giedion syndrome. Hum Mol Genet 1995;4(1):31-6. [ Links ]
2. Alanay Y, Lachman RS. A review of the principles of radiological assessment of skeletal dysplasias. J Clin Res Pediatr Endocrinol 2011;3(4):163-78. [ Links ]
3. Lüdecke HJ, Schaper J, Meinecke P, Momeni P, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet 2001;68(1):81-91. [ Links ]
4. Riedl S, Giedion A, Schweitzer K, Müllner-Eidenböck A, et al. Pronounced short stature in a girl with tricho-rhino-phalangeal syndrome II (TRPS II, Langer-Giedion syndrome) and growth hormone deficiency. Am J Med Genet A 2004;131(2):200-3. [ Links ]
5. Wells C, Spaggiari E, Malan V, Stirnemann JJ, et al. First fetal case of the 8q24.3 contiguous genes syndrome. Am J Med Genet A 2016;170A(1):239-42. [ Links ]
6. Momeni P, Glöckner G, Schmidt O, von Holtum D, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet 2000;24(1):71-4. [ Links ]
7. Li Q, Zhang Z, Yan Y, Xiao P, et al. Annular pancreas in Trichorhinophalangeal syndrome type II with 8q23.3-q24.12 interstitial deletion. Mol Cytogenet 2015;8:95. [ Links ]
8. Verheij JB, De Munnik SA, Dijkhuizen T, de Leeuw N, et al. An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur J Med Genet 2009;52(5):353-7. [ Links ]
9. Chen CP, Lin MH, Chen YY, Chern SR, et al. Prenatal diagnosis and array comparative genomic hybridization characterization of interstitial deletions of 8q23.3-q24.11 and 8q24.13 associated with Langer-Giedion syndrome, Cornelia de Lange syndrome and haploinsufficiency of TRPSI, RAD21 and Exil. Taiwan J Obstet Gynecol 2015;54(5):592-6. [ Links ]
10. Selenti N, Tzetis M, Braoudaki M, Gianikou K, et al. An interstitial deletion at 8q23.1-q24.12 associated with Langer-Giedion syndrome/ Trichorhinophalangeal syndrome (TRPS) type II and Cornelia de Lange syndrome 4. Mol Gytogenet 2015;8:64. [ Links ]
11. Parisi MS, Gazzerro E, Rydziel S, Canalis E. Expression and regulation of CCN genes in murine osteoblasts. Bone 2006;38(5):671-7. [ Links ]
12. Heath E, Tahri D, Andermarcher E, Schofield P, et al. Abnormal skeletal and cardiac development, cardiomyopathy, muscle atrophy and cataracts in mice with a targeted disruption of the Nov (Ccn3) gene. BMC Dev Biol 2008;8:18. [ Links ]
13. Giedion A. Phalangeal cone-shaped epiphyses of the hand: their natural history, diagnostic sensitivity, and specificity in cartilage hair hypoplasia and the trichorhinophalangeal syndromes I and II. Pediatr Radiol 1998;28(10):751-8. [ Links ]
14. Castriota-Scanderberg A, Dallapiccola B. Abnormal skeletal phenotypes: from simple signs to complex diagnoses. Berlin: Springer; 2005. [ Links ]

