










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.114 no.6 Buenos Aires dic. 2016
http://dx.doi.org/10.5546/aap.2016.e421
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2016.e421
Estudio de carcinoma medular de tiroides a partir de un caso índice
Study of Medullary Thyroid Carcinoma from a proband
Dra. Laura Morlán Herradora, Dr. Antonio de Arribaa, Dra. Gloria Miguela, Dra. Marta Ferrera y Dr. José I. Labartaa
a. Hospital Universitario Miguel Servet. Servicio de Endocrinología Pediátrica. Zaragoza. España.
Correspondencia: Dr. Antonio de Arriba, adearriba@salud.aragon.es
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 17-2-2016
Aceptado: 18-5-2016
RESUMEN
El carcinoma de tiroides es un tumor infrecuente; constituye menos del 1% de las neoplasias malignas en la población general y el 0,5%-3% en la edad pediátrica. Existen cuatro tipos: papilar (80%-90% de los casos), folicular (5%-10%), medular (5%) y anaplásico (2%-3%). En el tipo medular, el 80% son esporádicos, y un 20% se asocia a un síndrome hereditario que se divide, fundamentalmente, en tres grupos: neoplasia endócrina múltiple 1, neoplasia endócrina múltiple 2 y carcinoma medular de tiroides familiar. Las formas hereditarias se producen por una mutación en el protooncogén RET, localizado en el brazo largo del cromosoma 10.
Se presenta un caso de carcinoma medular de tiroides detectado a raíz de un estudio genético familiar con el propósito de resaltar la importancia del diagnóstico precoz y la intervención de equipos multidisciplinares expertos en esta patología para su manejo y seguimiento.
Palabras clave: Cáncer medular de tiroides; Técnicas genéticas; Diagnóstico precoz; Grupo de salud interdisciplinario.
ABSTRACT
Thyroid cancer is an uncommon type of cancer, accounting less than 1% of all cancers in adults, and 0.5-3% of all cancers in children. There are four different types: papillary carcinoma (80-90% of cases), follicular (5-10%), medullary (5%) and anaplastic cell (2-3%). Eighty per cent of cases of medullary thyroid cancer are sporadic, but 20% are associated with an inherited syndrome that is divided into three groups: multiple endocrine neoplasia type 1, multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma. The inherited forms are caused by a disruption in the RET oncogene, which is located in the long arm of chromosome 10.
A hereditary case of medullary thyroid carcinoma is presented. It was detected because of a familial genetic study. The purpose of the paper is emphasize the importance of the early diagnosis and the intervention of multidisciplinary teams of experts.
Key words: Medullary thyroid cancer; Genetic techniques; Early diagnosis; Interdisciplinary health team.
INTRODUCCIÓN
El carcinoma medular de tiroides (CMT) se origina en las células parafoliculares (células C) de la tiroides y es responsable del 2% de las neoplasias tiroideas malignas en la edad pediátrica. Se produce por una mutación en el protooncogén RET, localizado en el brazo largo del cromosoma 10, locus q11.2. Las formas hereditarias se dividen en neoplasia endócrina múltiple 1, neoplasia endócrina múltiple 2 y CMT familiar (CMTF).1 En los casos familiares, está relacionado con mutaciones en la línea germinal y, por tanto, puede ser transmitido a lo largo de sucesivas generaciones.
Una vez que el caso índice es diagnosticado en una familia, es de suma importancia identificar la mutación concreta del gen RET y estudiar a todos los miembros de la familia, puesto que la tiroidectomía previa a la diseminación del carcinoma es, actualmente, la mejor opción terapéutica.2
CASO CLÍNICO
Se presenta el caso de una paciente de 2 años remitida a la consulta de Endocrinología Pediátrica para estudio del protooncogén RET, debido a la detección de una mutación en dicho gen en una prima de la paciente. La paciente no presentaba antecedentes personales de interés, no existía consanguinidad entre sus padres, no había requerido intervenciones ni ingresos hospitalarios por patologías intercurrentes.
Antecedentes familiares (Figura 1): el caso índice de la familia era una prima hermana de la paciente, que fue diagnosticada con enfermedad de Hirschsprung, y se realizó un estudio genético de la mutación del protooncogén RET con resultado positivo, localizado en el cromosoma 10, locus q11.2. Se realizó una tiroidectomía profiláctica a los 2,5 años de edad con diagnóstico anatomopatológico de benignidad. A raíz de este caso, se realizó un estudio genético a los familiares y se encontraron 2 casos más. La madre del caso índice (tía paterna de la paciente) era portadora de dicha mutación, por lo que se realizó una tiroidectomía profiláctica a los 39 años de edad, con diagnóstico de CMT. El padre de la paciente que se presenta era también portador de la mutación y se realizó tiroidectomía, con diagnóstico de CMT (microcarcinoma medular en el lóbulo derecho e hiperplasia multinodular de células C bilateral). La hermana de la paciente no fue portadora de la mutación.
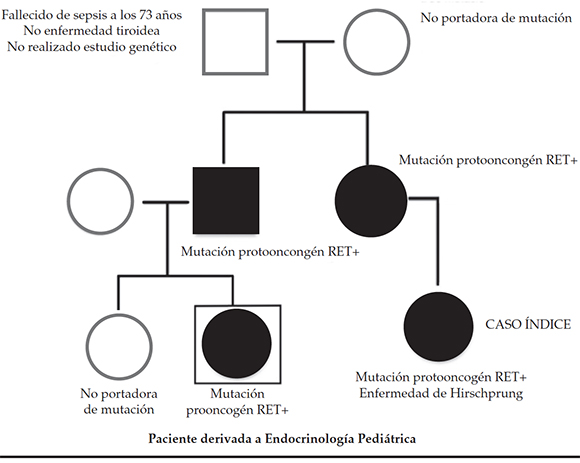
Figura 1. Árbol genealógico con estudios genéticos
Fallecido de sepsis a los 73 años No enfermedad tiroidea No realizado estudio genético
Diagnóstico y tratamiento: la paciente fue derivada para confirmar la mutación encontrada en este estudio familiar. Al momento de la exploración física, presentaba fenotipo y hábito sin alteraciones, con desarrollo psicomotor y ponderoestatural normal, sin presencia de bocio, con peso de 11,1 kg (-0,9 SDE) y talla de 84 cm (-0,88 SDE).
Se realizaron radiografía de tórax y ecografías abdominal y tiroidea, con resultados dentro de la normalidad. El análisis de sangre, que incluyó hemograma, hemostasia, bioquímica, estudio de la función tiroidea y del metabolismo del calcio, proteínas e inmunoglobulinas, fue normal. En el estudio genético, se confirmó la presencia de una mutación en el protooncogén RET (10q11.2) en el exón 10:c1859G>C, pcys620Ser en heterocigosis. Estos hallazgos se asociaron a CMTF y a síndrome de neoplasia endócrina múltiple 2A, por lo que se programó la tiroidectomía profiláctica, que se realizó a los 4 años de edad, sin incidencias, con diagnóstico anatomopatológico de hiperplasia microscópica multifocal.
Se inició el tratamiento tras la intervención con L-tiroxina y suplementos de calcio. Durante el seguimiento, ha mantenido niveles analíticos dentro de la normalidad, que incluyeron valores seriados de calcitonina (valor del último control < 2 pg/ml). Actualmente, han pasado 3 años desde la intervención y la paciente se encuentra estable y asintomática.
DISCUSIÓN
El CMT es un tumor neuroendócrino derivado de las células parafoliculares (células C), que son capaces de secretar sustancias hormonales, como la calcitonina y el antígeno carcinoembrionario (carcinoembryonic antigen; CEA, por sus siglas en inglés). Representa el 5%-10% de los tumores tiroideos malignos y se han descrito cuatro tipos según su forma de presentación: forma no familiar o esporádica (70%-80% del total), forma familiar aislada (5%-15% del CMT hereditario), formas familiares asociadas a otras alteraciones endócrinas (síndrome de la neoplasia endócrina múltiple -multiple endocrine neoplasia; MEN, por sus siglas en inglés- 2A y 2B) y formas familiares no asociadas a otras alteraciones endócrinas (CMTF). Dentro de las formas familiares, asociadas o no a otras alteraciones endócrinas, el MEN2A corresponde al 75% de las formas; el MEN2B, al 5%; y el CMTF, al 10%. Esta última corresponde al caso que se presenta.1
El síndrome MEN tipo 1 y 2 y el CMTF son trastornos autosómicos dominantes causados por mutaciones en el protooncogén RET, que afectan, fundamentalmente, a 4 tipos de tejidos derivados de las células de la cresta neural: células tiroideas parafoliculares tipo C, células paratiroideas, células cromafines de la médula adrenal y células del sistema nervioso autónomo mesentérico.3
El protooncogén RET se localiza en la región pericéntrica del brazo largo del cromosoma 10 (10q11.2) y está constituido por 21 exones que codifican el receptor RET, una proteína transmembrana citoplasmática de paso único, con actividad tirosina cinasa.4 Su activación produce hiperplasia de las células dianas in vivo con transformaciones que dan lugar a la formación de tumores. En el MEN2A, el 95% de las mutaciones ocurren en los exones 10, 11 y 14, mientras que, en el MEN2B, en el 95% de los casos, ocurren en el exón 16.5
El MEN2A se caracteriza por la presencia de CMT al comienzo de la edad adulta, feocromocitoma e hiperparatiroidismo. El MEN2B se caracteriza por CMT en la primera infancia y feocromocitomas asociadas a otras características no endocrinológicas, tales como enfermedad de Hirschprung, ganglioneuromatosis mucosa o hábito pseudomarfanoide.
Las recomendaciones de la Sociedad de Endocrinología Americana6 y de la Asociación de Tiroides Americana (ATA)4 sugieren el estudio del protoocongén RET en los siguientes casos: a) pacientes con CMT esporádico, b) miembros de primer grado de familias con CMTF, c) padres con niños que tienen el fenotipo clásico de MEN2B, d) pacientes con amiloidosis liquenoide cutánea, e) pacientes diagnosticados con feocromocitoma, f) niños con enfermedad de Hirschprung y alteraciones en el exón 10 del gen RET y g) adultos con MEN2A con síntomas sugestivos de enfermedad de Hirschprung.
La transformación maligna de las células C parafoliculares es un hecho muy precoz en la infancia y, además, la penetrancia para el CMTF es completa, por lo que el único tratamiento eficaz demostrado es la tiroidectomía profiláctica.7 Es por esto por lo que, ante la aparición de un caso índice con CMT, es imprescindible el estudio genético de los familiares, ya que permite un diagnóstico y tratamiento precoz.8
Actualmente, el estudio genético ha reemplazado las pruebas bioquímicas de determinación de niveles basales y luego de la estimulación con calcitonina, que se hacían en años anteriores como pesquisa de familiares de casos con CMT.9 Sin embargo, los niveles de calcitonina se relacionan con el tamaño tumoral; los niveles prequirúrgicos mayores de 500 pg/ml y su persistencia tras la cirugía son signos de mal pronóstico. Los pacientes con niveles de calcitonina mayores de 1000 pg/ml sin evidencia de CMT en el cuello se asocian con metástasis a distancia preferentemente a nivel hepático. Los niveles posquirúrgicos de calcitonina basal menores de 50 pg/ml pronostican una normalización posterior. Los niveles de calcitonina que descienden a menos de 10 pg/ml tras la cirugía indican mejor pronóstico, pero, incluso en los casos en los que los niveles de calcitonina son indetectables (menores de 2 pg/ml), no se puede asegurar la curación completa.10
Actualmente, la ATA recomienda la siguiente nomenclatura para clasificar a los pacientes afectos: se consideran pacientes de muy alto riesgo (highest risk, HST) aquellos con MEN2B y mutaciones en el codón M918T, y precisarán tiroidectomía total entre el mes y los seis primeros meses de vida, preferiblemente dentro del primer mes; se consideran pacientes de riesgo alto (high risk, H) aquellos con mutaciones en el codón RET C634 y A883F, que precisarán tiroidectomía total antes de los 5 años; finalmente, se agrupan el resto de mutaciones en una categoría de riesgo moderado (moderate risk, MOD), que incluye a pacientes con mutaciones en codones, tales como M918T, C634 y A883F.4 Estos últimos deberán individualizarse en cada caso y realizar la intervención teniendo en cuenta los niveles de calcitonina, preferentemente antes de los 10 años.
Tras la intervención, es preciso iniciar de inmediato el tratamiento con tiroxina para mantener una adecuado estado eutiroideo.11 Además, los niveles de calcio sérico deben ser monitorizados y, en caso de existir hipocalcemia sintomática, deberán administrarse suplementos de calcio y vitamina D.4
El seguimiento a largo plazo de estos pacientes consiste en mediciones anuales de calcitonina basal y de CEA.
La realización de la intervención por parte de un equipo multidisciplinar experto en esta patología para el manejo y seguimiento supone una tasa de éxito elevada que disminuye al mínimo las posibilidades de recidiva si la intervención se realiza dentro de los límites establecidos.12
1. Argente Oliver J, Soriano Guillén L. Patología de la glándula tiroides: hipotiroidismo, hipertiroidismo, nódulo de tiroides y cáncer de tiroides. En: Argente Oliver J, Soriano Guillén L, eds. Manual de Endocrinología Pediátrica 2a ed. Madrid: Ergon; 2014.Págs.168-170. [ Links ]
2. Kliegman R, Stanton B, St. Geme J, Schor N, et al. Carcinoma de tiroides. En Kliegman R, Stanton B, St. Geme J, Schor N, et al, eds. Nelson. Tratado de pediatría 19a ed. Madrid: Elsevier; 2011.Págs.1988. [ Links ]
3. Massoll N, Mazzaferri EL. Diagnosis and management of medullary thyroid carcinoma. Clin Lab Med 2004;24(1):49-83. [ Links ]
4. Wells SA Jr, Asa SL, Dralle H, Elisei R, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25(6):567-610. [ Links ]
5. Forga Llenas L. Genética del carcinoma medular de tiroides. Rev Endocrinol Nutr 2007;54(7):371-8. [ Links ]
6. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86(12):5658-71. [ Links ]
7. Machens A, Niccoli-Sire P, Hoegel J, Frank-Raue K, et al. Early malignant progression of hereditary medullary thyroid cancer. N Eng J Med 2003;349(16):1517-25. [ Links ]
8. Olivares Muñoz M, Juliá Masip MV, Oriola J, Martorell Sampol L, et al. La enfermedad de Hirschsprung y el carcinoma medular de tiroides: dos enfermedades en una alteración monogénica. Cir Pediátr 2012;25(2):87-90. [ Links ]
9. Rodríguez-Sánchez A, López-Menchero JC, Rodríguez-Arnao MD. Neoplasia endocrina múltiple: perspectiva pediátrica [Internet]. Madrid: Asociación Española de Cáncer de Tiroides. [Acceso: 19 de mayo de 2016]. Disponible en: http://www.aecat.net/wp-content/uploads/2014/01/neoplasia-endocrina-multiple-prespectiva-pediatrica.pdf.
10. Carles Genovés C, Martínez Sopena MJ, Rodríguez Arnau MD. Capítulo 31. Poliendocrinopatías autoinmunes. En Sociedad Española Endocrinología Pediátrica, eds. Guías diagnóstico-terapéuticas en Endocrinología Pediátrica. Valencia: Sociedad Española Endocrinología Pediátrica; 2004.Págs.1-10. [ Links ]
11. Cañizo A, Fanjul M, Cerdá J, Menárguez J, et al. ¿Es imprescindible la tiroidectomía profiláctica inmediata en el carcinoma medular de tiroides familiar? Cir Pediatr 2008;21(2):100-3. [ Links ]
12. Roldan Pérez S, Cabello Laureano R, Fernández-Pineda I, Aspiazu Salinas Y, et al. Hallazgos histológicos y seguimiento clínico de pacientes con MEN 2 tras tiroidectomía profiláctica. Cir Pediatr 2012;25(3):159-62. [ Links ]

