










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.115 no.1 Buenos Aires Feb. 2017
http://dx.doi.org/10.5546/aap.2017.e17
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2017.e17
Hemangiomatosis capilar pulmonar congénita en un recién nacido
Congenital pulmonary capillary hemangiomatosis in a newborn
Dra. Sandra L. Sposito Cavalloa, Dr. Luciano A. Macias Sobrinob, Dr. Luifer J. Marenco Altamarc y Dr. Andrés F. Mejía Alquichired
a. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Universidad de Buenos Aires, Unidad de Cuidados Intensivos Neonatales de la "Clínica de la Mujer", coordinadora del Área de Pediatría en la Universidad del Magdalena.
b. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan", Universidad de Buenos Aires, Unidad de Cuidados Intensivos Neonatales de la "Policlínica Ciénaga", docente de Pediatría de la Universidad del Magdalena.
c. Hospital Universitario Fernando Troconis, Universidad del Magdalena.
d. Hospital Universitario de Santander, Universidad del Magdalena.
Correspondencia: Dr. Luifer Marenco Altamar: elferlui@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 14-4-2016
Aceptado: 27-7-2016
RESUMEN
La hemangiomatosis capilar pulmonar es una entidad poco frecuente, caracterizada por la proliferación de capilares que infiltran paredes alveolares, septos interlobulillares, pleura e intersticio pulmonar, sin características de malignidad, con asociación casi constante a hipertensión pulmonar. Hasta el momento, solo se han reportado en la literatura, dos casos de presentación congénita; este es el tercer caso en un recién nacido y que no se asocia a hipertensión pulmonar. Esta se encuentra en la mayoría de los pacientes con dicha patología, con mayor incidencia entre los 20 y los 40 años de edad.
Se presenta a un recién nacido pretérmino de 36 semanas de gestación con dificultad respiratoria progresiva, que requirió asistencia ventilatoria mecánica por desaturaciones constantes en su evolución clínica, sin signos clínicos, radiológicos o ecográficos de hipertensión pulmonar.
Palabras clave: Hemangiomatosis capilar pulmonar; Congénita; Hipertensión pulmonar.
ABSTRACT
Pulmonary capillary hemangiomatosis is a rare entity characterized by the proliferation of capillaries into alveolar walls, interlobular septa, pleura and pulmonary interstitium, without malignant characteristics, with almost constant association with pulmonary hypertension. Until now two cases of congenital presentation have been reported in the literature. This is the third case in a newborn; he has not followed the usual pattern associated with pulmonary hypertension as occurs in most patients with this pathology; the highest incidence is among 20-40 years old.
We report a preterm newborn patient of 36 weeks of gestation with progressive respiratory distress requiring mechanical ventilation by constant desaturation during his clinical evolution without clinical, radiological or ultrasonographic signs of pulmonary hypertension.
Key words: Pulmonary capillary hemangiomatosis; Congenital; Pulmonary hypertension.
INTRODUCCIÓN
La hemangiomatosis capilar pulmonar (HCP), descrita por primera vez por Wagenvoort et al.1 en 1978, es una causa rara de hipertensión pulmonar (HTP), caracterizada por la proliferación de capilares que infiltran paredes alveolares, septos interlobulillares, pleura y tejido conjuntivo perivascular y peribronquial, y que, normalmente, no muestran características de malignidad.2 Puede presentarse en todos los grupos de edad con picos de incidencia entre los 20 y los 40 años sin predilección de sexo.3 Desde 1998, según Rich et al.,4 es considerada como entidad propia en la clasificación de la HTP de la Organización Mundial de la Salud junto con la HTP arterial idiopàtica y la enfermedad venooclusiva pulmonar (EVOP), y su distinción es importante porque los vasodilatadores pulmonares pueden ser perjudiciales en pacientes con HCP y EVOP.
La patogénesis de la HCP es desconocida. La HCP familiar ha sido descrita en tres hermanos, dos de ellos confirmados con la autopsia, que se reportaron con un patrón de herencia autosómico recesivo,5 y las mutaciones en el gen EIF2AK4, probablemente, causen HCP autosómica recesiva en los casos familiares y algunos no familiares.6 Solo se han reportado dos casos en la literatura en los que se presenta, de manera congénita, la HCP asociada a HTP.7 La presentación más común es la progresión lenta de HTP asociada a disnea de esfuerzo, cor pulmonale y, en última instancia, falla cardíaca derecha.8 Recientemente, se ha informado HCP sin HTP concomitante.9 También puede ocurrir con hemoptisis y derrame pleural.
Se presenta el caso de un recién nacido con HCP sin signos clínicos, radiológicos o ecográficos de HTP.
Presentación del caso
Recién nacido pretérmino tardío de 36 semanas de gestación, con peso adecuado de 3250 g, nacido por cesárea por causa materna.
Como antecedente, se trató de una madre de 21 años, sin consanguinidad con su pareja; embarazo controlado, con HTP inducida por el embarazo; con un período intergenésico corto de, aproximadamente, 12 meses. Su hijo anterior, también de sexo masculino, había fallecido en el período neonatal con diagnóstico de neumotórax bilateral y hemopericardio.
Luego de la adaptación neonatal, el paciente presentó signos de dificultad respiratoria progresiva, que requirió asistencia respiratoria mecánica. Mejoró la cianosis con saturación de 95%; frecuencia cardiaca: 155/min, temperatura: 36,2 °C. Se realizó una radiografía de tórax (véase la Figura 1), que evidenció opacidad pulmonar bilateral compatible con consumo de surfactante. Se descartó infección connatal, pero, dada la enfermedad pulmonar en el recién nacido, se administró surfactante pulmonar exógeno y se inició el tratamiento antibiótico con ampicilina y amikacina, inicialmente, durante 7 días, con realización de 2 hemocultivos antes y durante la antibioterapia, ambos negativos. Se logró optimizar la oxigenación con saturación de O2 mayor de 92% bajo ventilación mecánica convencional (VMC) con FIO2 100% y sedación. Se realizaron ecocardiogramas bidimensionales doppler color a la edad de 2, 3 y 4 meses, que reportaron presión de la arteria pulmonar de 35 mmHg, insuficiencia mitral mínima, ductus arterioso persistente, foramen oval permeable, buena contractilidad biventricular y ausencia de regurgitación tricúspide, sin evidencia de HTP. Evolucionó con hipoventilación generalizada y requerimiento de altos parámetros ventilatorios e intentos fallidos de extubación programada.

Figura 1: Radiografía de tórax del paciente recién nacido
Opacidad pulmonar bilateral compatible con síndrome de dificultad respiratoria (SDR); 8 espacios intercostales ventilados; tubo endotraqueal normoinserto.
Ante la evidencia de enfermedad pulmonar persistente corroborada por opacidad pulmonar bilateral con infiltrados difusos, al mes de edad, se realizó una tomografía axial computarizada (TAC) de tórax simple (véase la Figura 2), que reportó volúmenes pulmonares normales con aumento del intersticio pulmonar, consolidaciones pulmonares bibasales asociadas a broncograma aéreo con patrón de vidrio esmerilado, que orientó a enfermedad intersticial difusa, proceso infeccioso pulmonar o hemorragia alveolar difusa. De manera concomitante, presentó hepatomegalia palpable e ictericia con hiperbilirrubinemia directa, médicamente tratada, asumida como secundaria a nutrición parenteral prolongada.

Figura 2. Tomografía axial computarizada simple de tórax
Volúmenes pulmonares normales con aumento del intersticio pulmonar.
El paciente evolucionó con alteraciones tanto en la ventilación como oxigenación, con gases arteriales con aumento de CO2, hipoxemia y alcalosis respiratoria compensatoria, manifestaciones radiográficas con un patrón heterogéneo de áreas más ventiladas y zonas de atelectasias, con compromiso del parénquima pulmonar.
Ante la necesidad de ventilación mecánica prolongada con altos requerimientos de oxígeno, episodios de broncoespasmo e hipersecreción de moco, descartando enfermedad cardíaca, sin mejoría ante el tratamiento con surfactante y ciclos de antibióticos, se decidió realizar una toracotomía y biopsia de pulmón. Fueron enviadas a Patología, quienes realizaron el diagnóstico. La anatomía patológica (Figuras 3 y 4) informó tejido pulmonar con engrosamiento septal difuso y de grado variable por proliferación de capilares, que se confirmó con el marcador cd 34 y no se evidenció fibrosis intersticial a la coloración de tricromo. Los tabiques alveolares contenían más de una hilera de capilares, que mostraron congestión y revestimiento endotelial. A nivel bronquiolar, no se observaron cambios. La afectación arterial era mínima y consistía en un ligero engrosamiento de la capa media. La coloración PAS (periodic acid-Schiff) no demostró depósitos de glicógeno y la coloración de Ziehl-Neelsen y Grocott fueron negativas para bacterias ácido-alcohol resistentes (BAAR) y hongos. Se diagnosticó HCP congénita.
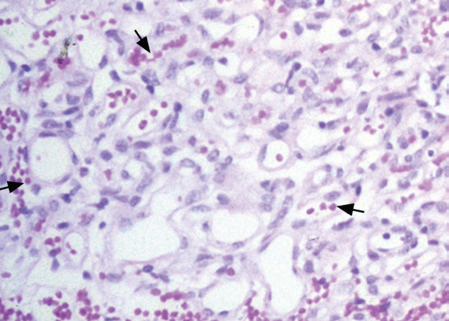
Figura 3. Biopsia pulmonar
Coloración de hematoxilina-eosina (HE) a 40X que muestra (flechas) un engrosamiento a nivel intersticial secundario a proliferación anormal de capilares en la arquitectura pulmonar, característica de la HCP.

Figura 4. Biopsia pulmonar
Marcador CD34 positivo que resalta la proliferación de capilares a nivel intersticial (flechas), con vasos sanguíneos que dificultan la dinámica ventilatoria. Técnica de inmunoperoxidasa a 40X.
Debido a la deficiencia respiratoria secundaria a esta entidad sin tratamiento definitivo, se inició tratamiento médico sintomático con espironolactona, furosemida, salbutamol asociado a bromuro de ipratropio, budesonida inhalatoria según síntomas. Se indicó apoyo nutricional a través de gastrostomía, tratamiento de intercurrencias y medidas paliativas para generar bienestar.
El bienestar físico del paciente en su evolución se vio alterado no solo por su patología crónica de base, sino también por enfermedades concomitantes, como hernia inguinal derecha, que luego se encarceló y fue resuelta quirúrgicamente, así como cuadro de otitis media aguda y neumonías asociadas a ventilación mecánica. A pesar de que no se les hizo ningún tipo de estudio genético a los padres, se explicó a la madre el riesgo en un futuro embarazo.
Finalmente, el paciente falleció a los 8 meses de vida producto de insuficiencia respiratoria refractaria al tratamiento.
DISCUSIÓN
El diagnóstico definitivo de esta entidad solo puede establecerse con precisión por examen histológico; en la mayoría de los casos, se hace en la autopsia o como resultado de neumonectomía durante la vida.10,11 La proliferación de capilares alveolares de paredes finas con infiltración de las paredes de vasos sanguíneos pulmonares septales, vías respiratorias y la pleura, en ausencia de la esclerosis venosa marcada, es una característica histopatológica distintiva.8 En la radiografía de tórax, se muestra un patrón micronodular difuso.12 El trasplante ortotópico de corazón-pulmón o de doble pulmón es considerado como la única alternativa terapéutica,12,13 aunque algunos informes de caso parecen sugerir éxito terapéutico con interferón e inhibidores de angiogénesis, tales como la doxiciclina.14 El pronóstico para HCP es pobre y la mayoría de los pacientes mueren de la enfermedad dentro de 1-5 años después de la aparición de los síntomas.
Hubo dos casos presentados en la literatura en los que la HCP se reportó de manera congénita: uno de ellos fue un gemelo mortinato con agenesia de ambos riñones; el segundo tuvo HTP, insuficiencia mitral y una miocardiopatía hipertrófica asociada.7 Nuestro reporte es el tercer caso de presentación congénita, que muestra un comportamiento diferente en ausencia total de HTP.
Aunque el paciente no presentaba HTP, sí mostró cuadros recurrentes de desaturaciones asociados a dificultad respiratoria, presentes desde su nacimiento con episodios de broncoespasmos e hipersecreción mucosa, que respondía con una mejoría leve y transitoria en cada una de sus exacerbaciones al tratamiento paliativo, pues la única alternativa terapéutica eficaz, que es el trasplante ortotópico, no está disponible en el país.
Debido a su baja incidencia en todos los grupos de edades, especialmente en recién nacidos, se sabe muy poco acerca de la enfermedad y de sus diferentes tipos de presentación al ocurrir o no con HTP, en ausencia de estadísticas que revelen la supervivencia y manejo pertinente al enfrentarse a estos casos, que pueden ser solo diagnosticados bajo un estudio histológico de biopsia de pulmón, cuando se hayan agotado todas las opciones diagnósticas. Dentro de los hallazgos radiológicos, aunque se ha relacionado el patrón de vidrio esmerilado con HCP, en todos los grupos de edades, solo es efectivo como orientación diagnóstica para pensar en una enfermedad pulmonar que afecta el intersticio, como HCP, neumonía crónica de la infancia, neumonía atípica, otras entidades limitadas a hemorragia alveolar difusa y EVOP. Todas con muy baja incidencia en la infancia.
Agradecimientos
Se hace mención especial a la Dra. Paulina Ojeda de León, médica patóloga pulmonar del "Jackson Memorial Hospital of Miami", quien labora en la ciudad de Bogotá, Colombia, por hacer el diagnóstico patológico de esta entidad tan poco común y por sus importantes aportes en la realización de esta presentación de caso.
1. Wagenvoort CA, Beetstra A, Spijker J. Capillary haemangiomatosis of the lungs. Histopathology 1978;2(6): 401-6. [ Links ]
2. Travis WD, Colby TV, Koss MN, Rosado-de-Christenson ML, et al. Pulmonary capillary hemangiomatosis. Chapter 15. In Non-Neoplastic Disorders of the Lower Respiratory Tract. Washington: American Registry of Pathology; 2002:788-9. [ Links ]
3. Almagro P, Julià J, Sanjaume M, González G, et al. Pulmonary capillary hemangiomatosis associated with primary pulmonary hypertension: report of 2 new cases and review of 35 cases from the literature. Medicine (Baltimore) 2002;81(6):417-24. [ Links ]
4. Rich S, Rubin LJ, Abenhail L, eds. Executive summary from the World Symposium on Primary Pulmonary Hypertension;1998 September 6-10; Evian, France. Geneva: World Health Organization; 1998. [ Links ]
5. Langleben D, Heneghan JM, Batten AP, Wang NS, et al. Familial pulmonary capillary hemangiomatosis resulting in primary pulmonary hypertension. Ann Intern Med 1988;109(2):106-9. [ Links ]
6. Best DH, Sumner KL, Austin ED, Chung WK, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014;145(2):231-6. [ Links ]
7. Oviedo A, Abramson LP, Worthington R, Dainauskas JR, et al. Congenital pulmonary capillary hemangiomatosis: Report of two cases and review of the literature. Pediatr Pulmonol 2003;36(3):253-6. [ Links ]
8. Eltorky MA, Headley AS, Winer-Muram H, Garrett HE, Jr, et al. Pulmonary capillary hemangiomatosis: a clinicopathologic review. Ann Thorac Surg 1994;57(3):772-6. [ Links ]
9. Umezu H, Naito M, Yagisawa K, Hattori A, et al. An autopsy case of pulmonary capillary hemangiomatosis without evidence of pulmonary hypertension. Virchows Arch 2001;439(4):586-92. [ Links ]
10. Domingo C, Encabo B, Roig J, López D, et al. Pulmonary capillary hemangiomatosis: report of a case and review of the literature. Respiration 1992;59(3):178-80. [ Links ]
11. Faber CN, Yousem SA, Dauber JH, Griffith BP, et al. Pulmonary capillary hemangiomatosis. A report of three cases and a review of the literature. Am J Resp Dis 1989;140(3):808-13. [ Links ]
12. Tron V, Magee F, Wright JL, Colby T, et al. Pulmonary capillary hemangiomatosis. Hum Pathol 1986;17(11):1144-50. [ Links ]
13. Magee F, Wright JL, Kay J, Peretz D, et al. Pulmonary capillary hemangiomatosis. Am Rev Respir Dis 1985;132(4):922-5. [ Links ]
14. White CW, Sondheimer HM, Crouch EC, Wilson H, et al. Treatment of pulmonary hemangiomatosis with recombinant interferon alfa-2a. N Engl J Med 1989; 320(18):1197-200. [ Links ]

