










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.115 no.3 Buenos Aires June 2017
http://dx.doi.org/10.5546/aap.2017.e170
PRESENTACIÓN CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2017.e170
Síndrome de Cornelia de Lange y deficiencias hormonales pituitáricas múltiples, una asociación inusual. Caso clínico
Cornelia de Lange Syndrome and multiple hormonal deficiency, an unusual association. Clinical case
Dr. Víctor M. Mora-Bautistaa, Dr. Víctor Mendoza-Rojasb,d y Dr. Gustavo A. Contreras-Garcíac,d
a. Departamento de Pediatría, Universidad Industrial de Santander, Bucaramanga. Santander, Colombia.
b. Departamento de Pediatría, Escuela de Medicina, Universidad Industrial de Santander.
c. Escuela de Medicina, Universidad Industrial de Santander.
d. Hospital Universitario de Santander.
Correspondencia: Dr. Víctor Mendoza-Rojas, victormr@uis.edu.co
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 17-9-2016
Aceptado: 26-12-2016
RESUMEN
El síndrome de Cornelia de Lange es una enfermedad genética caracterizada por rasgos faciales distintivos, falla de medro, microcefalia y varias malformaciones asociadas. Sus principales alteraciones endocrinológicas son las anomalías genitales. Se presenta un adolescente de 13 años, tratado por neumonía aspirativa complicada y que presentaba el fenotipo del síndrome de Cornelia de Lange, que incluía retraso global del desarrollo, trastorno de succión-deglución, talla baja y alteración del desarrollo sexual. Su edad ósea era muy retrasada, por lo que se realizó un estudio endocrinológico completo. Se le diagnosticaron hipotiroidismo central, deficiencia de la hormona de crecimiento y deficiencia de hormona luteotrópica y folículoestimulante, compatibles con el diagnóstico de deficiencias hormonales pituitáricas múltiples. Tuvo cortisol basal, hormona adrenocorticotrópica y prolactina normales. Recibió suplencia hormonal tiroidea. Es inusual la asociación de este síndrome con deficiencias hormonales pituitáricas múltiples. Se sugiere la evaluación de los distintos ejes endócrinos en estos pacientes.
Palabras clave: Síndrome de Cornelia de Lange; Talla baja; Criptorquidismo; Malformaciones congénitas; Deficiencias hormonales pituitáricas múltiples.
ABSTRACT
Cornelia de Lange syndrome is a genetic disease characterized by distinctive facial features, failure to thrive, microcephaly and several malformations associated. Its main endocrinological features are anomalies of the genitalia. We present a 13-year-old boy, who suffered from complicated aspiration pneumonia and showed Cornelia de Lange syndrome phenotype, with global developmental delay, suction-swallowing abnormalities, short stature and abnormal genitalia associated. His bone age was delayed, so he underwent full endocrinological panel.
Central hypothyroidism, growth hormone deficiency and low luteinizing hormone-follicle-stimulating hormone levels were observed and multiple pituitary hormone deficiencies diagnosis was made. Basal cortisol, adrenocorticotropic hormone and prolactin levels were normal. He received thyroid hormonal substitution. Multiple pituitary hormone deficiencies are an unusual feature of De Lange syndrome. We suggest evaluating all different endocrine axes in these patients.
Key words: Cornelia de Lange syndrome; Short stature; Cryptorchidism; Congenital malformations; Multiple pituitary hormone deficiencies.
INTRODUCCIÓN
El síndrome de Cornelia de Lange (SCdL) (OMIM #122470, #300590, #610759, #614701 y #300882) es una enfermedad genética con heterogeneidad de loci y variabilidad de expresión, caracterizado por rasgos faciales distintivos, falla de medro, microcefalia y varias malformaciones asociadas. Las malformaciones genitourinarias descritas principalmente son micropene, hipospadia y criptorquidia. Sin embargo, aún es de diagnóstico clínico, como en 1933, cuando fue descrito por la pediatra de quien lleva su nombre. Hay quienes atribuyen la descripción inicial al Dr. Brachmann en 1916. Su incidencia varía entre 1:10 000 y 1:30 000 nacidos vivos.1-6 En Colombia, no hay datos precisos.7
La asociación de deficiencias hormonales pituitáricas múltiples con el SCdL se presumía como un componente del síndrome en sus primeras descripciones, pero ahora poco se menciona como parte del síndrome.8,9 A pesar de esto, hay descripciones anatomopatológicas de compromiso de glándulas adrenales, paratiroides, tiroides y quistes en la adenohipófisis.10
OBJETIVO
Se presenta la historia médica de un paciente con SCdL en el que coexisten las deficiencias hormonales pituitáricas múltiples. Con este caso, se desea alertar sobre la necesidad de vigilancia de la función hormonal en el manejo de estos pacientes.
CASO CLÍNICO
Adolescente de 13 años, proveniente de una zona rural distante, quien estuvo en tratamiento en el Hospital Universitario de Santander durante 10 días por neumonía aspirativa complicada, con 2 semanas previas de síntomas. Además, presentaba anemia crónica multifactorial (carencial y secundaria a infección), junto con falla renal aguda prerrenal a causa de deshidratación por inapetencia, las cuales se resolvieron durante la estancia.
Fue llamativo que presentaba un grupo de malformaciones múltiples asociadas sin clasificar, con retraso global del desarrollo y trastorno de succión-deglución, que condicionaron múltiples episodios previos de neumonía aspirativa.
En la familia, solo se conocía un caso de síndrome de Down y no había otros familiares con malformaciones. Hijo de la tercera de cuatro gestaciones de la misma unión, con controles prenatales incompletos y parto domiciliario hacia las 30 semanas debido a dificultades geográficas de su lugar de residencia. No había consanguinidad entre los padres. Fue evaluado médicamente por primera vez a los 15 días de vida (también por limitaciones geográficas y económicas) y estuvo hospitalizado por sepsis neonatal. Adquirió sostén cefálico a los 2 años, roló a los 2 años, logró la sedestación con apoyo a los 5 años y no gateaba ni caminaba; solo emitía sonidos guturales.
Al momento del examen físico de ingreso, se observó talla baja proporcionada, inferior a -4 desvíos estándar (DE) -peso de 11,9 kg y talla de 99 cm, perímetro cefálico (PC) de 44 cm-, sinofris, cejas arqueadas, tricomegalia, narinas pequeñas y levemente antevertidas, puente nasal deprimido, filtrum plano, labios delgados y comisuras labiales hacia abajo (Figura 1). También se halló paladar duro con hendidura ósea posterior e integridad mucosa, diastema y protrusión dental, cuello corto, diámetro anteroposterior del tórax aumentado y trece costillas bilaterales (dato corroborado por radiografía; véase la Figura 2). Tenía un ombligo pequeño, genitales anormales con criptorquidia bilateral, escroto hipopigmentado hipoplásico, sin rugosidades y micropene con uretra normoinserta (Figura 3). Al momento del examen perineal, se observó un ano de inserción anterior. Presentaba braquidactilia con hipocratismo; además, tenía un pliegue palmar único, clinodactilia, ausencia de pliegues interfalángicos proximales en los dedos de las manos con sinfalangismo y 1er metacarpiano corto con localización proximal del pulgar. Sus pies eran pequeños, con sindactilia entre el segundo y el tercer dedo. La piel era seca, descamativa, con palidez y cutis marmorata. Exhibía conductas autolesivas ocasionales y resistencia al cambio. Fue evaluado por genética y endocrinología pediátrica, y se estableció el diagnóstico de SCdL.
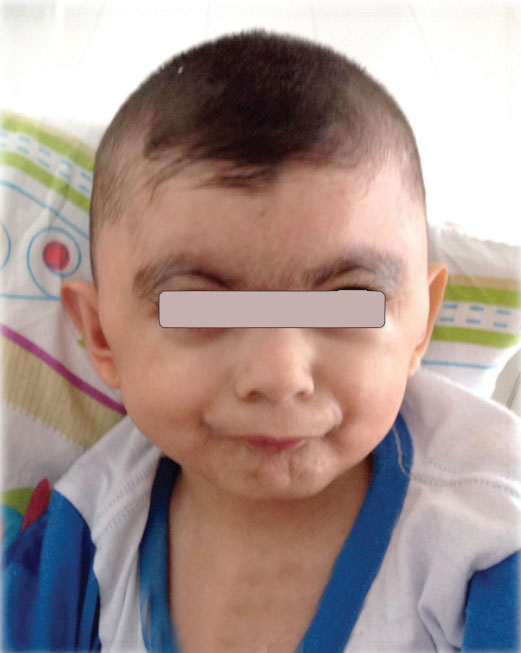
Figura 1. Características faciales
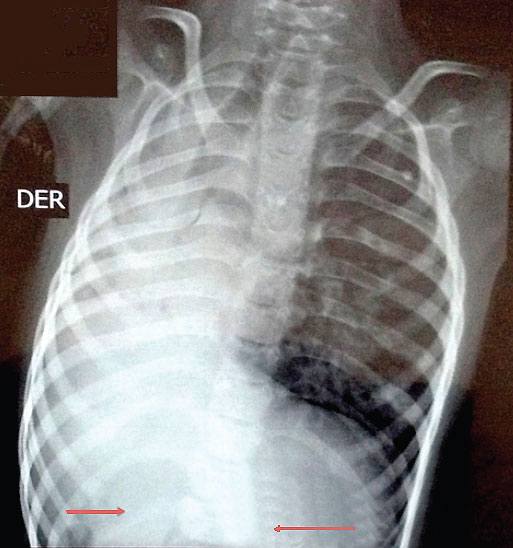
Figura 2. Radiografía de tórax con costillas supernumerarias

Figura 3. Hipogonadismo genital con micropene y criptorquidia
Dentro del estudio de su talla baja y alteración del desarrollo sexual, se halló una edad ósea de 4 años y medio según Greulich y Pyle. Se evaluaron todos los ejes hipofisarios (Tabla 1). Se encontró hipotiroidismo central, para el cual se inició suplencia con 70 mcg/m2/día de levotiroxina. Las gonadotrofinas fueron reportadas en el límite inferior, aunque debieron ser interpretadas con cautela por ser prepuberales; podrían sugerir hipogonadismo hipogonadotrópico, pero, por su edad, no estaba indicado iniciar un tratamiento hormonal. Los reportes de electrolitos fueron normales y no presentaba síndrome poliúrico-polidípsico. Ya había sido evaluado por oftalmología y otorrinolaringología hacia los dos años en forma ambulatoria, sin hallar anormalidades, sin nuevo seguimiento hasta la fecha.
Tabla 1. Ejes hormonales evaluados en el paciente

Se hizo una ecografía testicular, con criptorquidia derecha (atrófico y sin flujo) e imposibilidad de visualizar el testículo izquierdo. No fue posible realizar la resonancia magnética nuclear (RMN) hipofisaria debido a la negativa de los padres a realizar más estudios.
La suplencia tiroidea y la fisioterapia (sesiones diarias de terapia física y ocupacional) mejoraron su nivel de actividad física, la capacidad de interacción con el medio y desapareció su piel seca y descamativa en el transcurso de las dos primeras semanas. Egresó luego de 20 días de estancia y recibió 25 μg diarios de levotiroxina e indicación de seguimiento ambulatorio en 4 semanas. Desafortunadamente, no asistió a las consultas, a pesar del esfuerzo del servicio social del Hospital para su seguimiento integral.
DISCUSIÓN
Los avances en el SCdL han permitido establecer mejor su etiología. Se dieron los mayores saltos desde 2004 con la identificación de las alteraciones del gen NIPBL. Desde entonces, se han identificado mutaciones que afectan la síntesis de cohesinas (NIPBL, SMC1A, SMC3, RAD21 y HDAC8). Estas proteínas son importantes en la separación de cromátides antes de la mitosis, lo que explica que haya alteraciones en múltiples sistemas orgánicos.11 Existen cinco grupos fenotípicos, uno por cada gen.6 A pesar de esta genotipificación, el diagnóstico es, fundamentalmente, clínico.
En el caso presentado, la familia no había podido acceder a la prueba genética ni dio su consentimiento para intentar realizarla durante la última estancia hospitalaria.
Aunque hay un perfil clásico bastante heterogéneo en la enfermedad, características tales como predisposición a neumonías, trombocitopenia y sordera neurosensorial son percibidas como atípicas (Tabla 2). Las alteraciones endócrinas se consideran aún más inusuales, aunque, inicialmente, se estimaban parte del síndrome. Las más frecuentes reportadas (hasta 40%) son la hipospadia, el micropene y la criptorquidia.1,5,10,12
Tabla 2. Fenotipo y características clínicas del síndrome de Cornelia de Lange

La complejidad y gravedad del compromiso endócrino es variable, y se comunican casos aislados de hipoglucemias neonatales, hipotiroidismo, hipogonadismo y, menos frecuentemente, deficiencias hormonales pituitáricas múltiples, los cuales podrían explicarse por hipoplasia de la hipófisis, de las adrenales o de la tiroides.8,9 Se ha reportado que el hipogonadismo es variable, y llegan a presentarse desarrollo puberal y casos de embarazos viables.9 Además, no suelen hallarse alteraciones anatómicas de la hipófisis, sino un problema funcional. Incluso en estos pacientes, radiológicamente, solo se han informado tumores, quistes, cambios en la materia blanca, atrofia cerebral, hipoplasia cerebelosa y ventriculomegalia.13
CONCLUSIONES
Pocos casos se han descrito en la literatura que relacionen el SCdL con deficiencias hormonales múltiples, como fue demostrado en el paciente. El manejo de estos pacientes debe incluir pediatría, endocrinología, otorrinolaringología, gastroenterología, urología, odontología, neuropediatría, genética, fisiatría, ortopedia y un especialista en neurorehabilitación. Según la complejidad, pueden requerirse otras especialidades médicas, así como enfermería, psicología y asistencia social.14 Desafortunadamente, fuera de los problemas de accesibilidad y aseguramiento, existen aún barreras socioculturales en la atención de estos niños, que demandan un esfuerzo desde los primeros niveles de atención en salud.
1. Gil MC, Ribate MP, Ramos FJ. Síndrome de Cornelia de Lange. Protoc Diagn Ter Pediatr 2010;1:1-12. [ Links ]
2. Tekin M. Cornelia De Lange Syndrome [Internet]. Medscape; 2015. [Consulta: 9 de enero de 2017]. Disponible en: http://emedicine.medscape.com/article/942792-overview.
3. Orphanet. Síndrome de Cornelia de Lange [Internet]. París: Orphanet; 2009. [Consulta: 9 de enero de 2017]. Disponible en: http://www.orpha.net/consor/cgi-bin/OC_Exp. php?Lng=ES&Expert=199.
4. Kousseff BG, Newkirk P, Root AW. Brachmann-de Lange syndrome. 1994 update. Arch Pediatr Adolesc Med 1994;148(7):749-55. [ Links ]
5. Mannini L, Cucco F, Quarantotti V, Krantz ID, et al. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum Mutat 2013;34(12):1589-96. [ Links ]
6. Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet 2009;76(4):303-14. [ Links ]
7. Montes ML, Saldarriaga W, Isaza C. Descripción de un caso de síndrome de Cornelia de Lange. Aporte para un mejor diagnóstico pre- y post-natal. Colomb Med 2006;37(4):323-7. [ Links ]
8. Tonini G, Marinoni S. Neonatal-onset panhypopituitarism in a girl with Brachmann-De Lange syndrome. Am J Med Genet 1990;36(1):102-3. [ Links ]
9. Schwartz ID, Schwartz KJ, Kousseff BG, Bercu BB, et al. Endocrinopathies in Cornelia de Lange syndrome. J Pediat 1990;117(6):920-3. [ Links ]
10. Schrier SA, Sherer I, Deardoff MA, Clark D, et al. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet A 2011;155A(12):3007-24. [ Links ]
11. Brooker AS, Berkowitz KM. The roles of cohesins in mitosis, meiosis, and human health and disease. Methods Mol Biol 2014;1170:229-66. [ Links ]
12. Cheung K, Upton J. Cornelia de Lange Syndrome. J Hand Surg Am 2015;40(12):2501-3. [ Links ]
13. Whitehead MT, Nagaraj UD, Pearl PL. Neuroimaging features of Cornelia de Lange syndrome. Pediatr Radiol 2015;45(8):1198-205. [ Links ]
14. Mikotajewska E. Interdisciplinary therapy in Cornelia de Lange syndrome - review of the literature. Adv Clin Exp Med 2013;22(4):571-7. [ Links ]

