










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.115 no.3 Buenos Aires jun. 2017
http://dx.doi.org/10.5546/aap.2017.e183
PRESENTACIÓN CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2017.e183
Trisomía 18 en mosaico. Serie de casos
Mosaic trisomy 18. Series of cases
Prof. Francisco Cammarata-Scalisia, Prof. María A. Lacruz-Rengelb, Lic. Dianora Araquea, Prof. Gloria Da Silvaa, Prof. Andrea Avendañoa, Dr. Michele Calleac, Dra. Frances Stockb, Dra. Yudith Guerreroe, Br. Eliomar Aguilarf, Br. María J. Lacruzf y Br. Jesús Sulbaranf
a. Unidad de Genética Médica. Departamento de Puericultura y Pediatría. Universidad de Los Andes. Mérida, Venezuela.
b. Servicio de Neuropediatría. Departamento de Puericultura y Pediatría. Universidad de Los Andes. Mérida, Venezuela.
c. Unit of Dentistry. Bambino Gesù Children's Hospital -IRCCS. Rome, Italy.
d. Unidad de Oncología Pediátrica. Instituto Autónomo Hospital Universitario de Los Andes. Mérida, Venezuela.
e. Hospital Tulio Febres Cordero. La Azulita, Mérida, Venezuela.
f. Facultad de Medicina. Universidad de Los Andes. Mérida, Venezuela.
Correspondencia: Prof. Francisco Cammarata-Scalisi, francocammarata19@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 25-10-2016
Aceptado: 11-2-2017
RESUMEN
El síndrome de la trisomía 18 es un trastorno clínico y genético, el cual presenta un cromosoma 18 extra completo en cada célula, variante que se denomina trisomía libre. Además, puede ocurrir en la forma parcial y mosaico. Clínicamente, se caracteriza por retardo del crecimiento intrauterino, del desarrollo psicomotor y mental, hallazgos craneofaciales característicos, cardiopatía congénita, pelvis hipoplásica, manos empuñadas y pies en mecedora, entre otros. La trisomía 18 en mosaico se presenta cuando células con trisomía del cromosoma 18 y líneas celulares normales existen en un mismo individuo, y corresponde al 5% de los casos. Los hallazgos fenotípicos son muy variables y no se evidencia una correlación entre el porcentaje de células trisómicas y los hallazgos encontrados. El objetivo de este informe es presentar una serie de cinco casos de trisomía 18 en mosaico. Se hace énfasis en los aspectos clínicos con la finalidad de orientar una adecuada atención médica interdisciplinaria y brindar un oportuno asesoramiento genético.
Palabras clave: Trisomía 18; Mosaicismo; Asesoramiento genético.
ABSTRACT
Trisomy 18 syndrome (T18) is a clinical and genetic disorder, which has a full extra chromosome 18 in each cell, variant that is called free trisomy. In addition, it can occur in partial and mosaic form. It is characterized by intrauterine growth restriction, psychomotor and mental retardation, characteristic craniofacial findings, congenital heart disease, hypoplastic pelvis, clenched hand and rocker-bottom foot, among others. The mosaic T18 occurs when cells with T18 and normal cell lines exist in the same individual and correspond to 5% of cases.
The phenotypic findings are highly variable and no correlation was evident between the percentage of trisomic cells and the findings found. The aim of this report is to present a series of five cases of mosaic T18 with emphasis on clinical aspects in order to guide an interdisciplinary adequate medical care and provide timely genetic counseling.
Key words: Trisomy 18; Mosaicism; Genetic counseling.
INTRODUCCIÓN
El síndrome de la trisomía 18 (T18) o síndrome de Edwards es un trastorno clínico y genético que se caracteriza por la presencia de un cromosoma 18 extra completo en cada célula.1 La trisomía libre representa alrededor de 94% de los casos.1,2 Presenta una incidencia de 1 en 6000-8000 nacimientos.1 Se puede evidenciar retardo del crecimiento intrauterino (RCIU), del desarrollo psicomotor y mental, malformaciones en el sistema nervioso central, esternón corto, cardiopatía congénita, alteraciones urogenitales, pelvis hipoplásica, manos con dedos superpuestos, pies en mecedora e hipoplasia ungueal, entre otros. Aproximadamente, 90% de los pacientes fallecen en el primer año.1-3
Además, se han presentado casos de T18 en forma parcial y mosaico.1 La T18 en mosaico se presenta cuando células con trisomía del cromosoma 18 y líneas celulares normales existen en un mismo individuo, y corresponde al 5% de los casos. Los hallazgos fenotípicos son variables. No obstante, no se evidencia una correlación entre el porcentaje de células trisómicas en leucocitos o fibroblastos y los hallazgos clínicos encontrados.1,2
El objetivo es presentar una serie de cinco casos, en el período de 2011-2015, de T18 en mosaico. Se hace énfasis en los hallazgos clínicos con la finalidad de orientar una atención médica interdisciplinaria y brindar un oportuno asesoramiento genético familiar.
DESCRIPCIÓN DE LOS CASOS
Caso 1
Recién nacido femenino evaluado por presentar RCIU grave y múltiples malformaciones congénitas (Figura 1). Madre de 21 años de edad, primera gesta, embarazo gemelar bicorial y biamniótico, complicado con infección en las vías urinarias y preeclampsia grave. Nacimiento por cesárea a las 37 semanas. Requirió de reanimación cardiopulmonar avanzada por depresión neonatal grave, que ameritó ventilación mecánica. El peso al nacer fue de 1400 g (P < 3) SDE -4,6 y la talla, de 40 cm (P < 3) SDE -7,2.
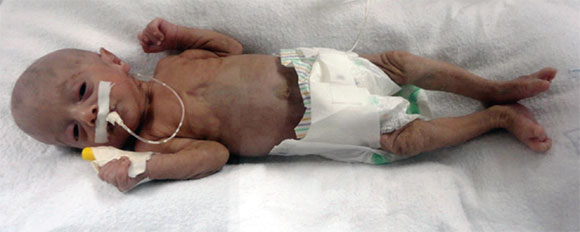
Figura 1. Se evidencia microdolicocefalia, facies progeroide, cejas escasas, ptosis palpebrai bilateral, narinas antevertidas con punta nasal bulbosa y, en las extremidades, las manos empuñadas con el segundo dedo sobre el tercero y el quinto sobre el cuarto
La variabilidad fenotípica se exhibe en la Tabla 1. La ecografía transfontanelar realizada al segundo día de nacida mostró cisterna magna y cavum del septum pellucidum persistente. La ecocardiografia transtorácica realizada al sexto día evidenció una comunicación interventricular de 3,4 mm con cortocircuito de izquierda a derecha tratada con furosemida y espironolactona. La ecografía renal realizada al mes de edad mostró ureteropielectasia derecha de 1,93 cm.
Tabla 1. Hallazgos clínicos encontrados en los pacientes con trisomía 18 en mosaico

La evolución fue desfavorable, y presentó endocarditis infecciosa por Staphylococcus aureus tratada con penicilina G cristalina.
Posteriormente, status convulsivo tratado con fenitoína, insuficiencia respiratoria global y bloqueo auriculoventricular completo de segundo grado Mobitz II. Anemia moderada con tiempos de coagulación prolongados, hipoglicemia, hipoproteinemia, desequilibrio ácido-base y de electrólitos caracterizado por acidosis mixta, hipernatremia e hipopotasemia. Presentó un paro cardiorrespiratorio y falleció a los tres meses y 22 días.
Caso 2
Recién nacido femenino evaluado por presentar RCIU grave y cardiopatia congènita. Madre de 43 años de edad, segunda gesta, embarazo complicado con hipertensión arterial en la última semana e hipomotilidad fetal. Nacimiento por cesárea a las 38 semanas, por circular de cordón umbilical y presentación podálica. Requirió reanimación cardiopulmonar avanzada por depresión neonatal moderada. El peso al nacer fue de 2200 g (P < 3) SDE -2,5 y la talla, de 44 cm (P < 3) SDE -4,1.
La ecocardiografia transtorácica al día de nacida evidenció una comunicación interauricular tipo ostium secundum de 6,4 mm, comunicación interventricular perimembranosa de 6,7 mm, persistencia de ductus arterioso de 4 mm e insuficiencia aórtica leve. Se inició un tratamiento con ibuprofeno y furosemida; posteriormente, furosemida, espironolactona, captopril y beta metildigoxina. La ecografía transfontanelar realizada al mes de edad mostró una asimetría de los ventrículos laterales que podía corresponder a una variante anatómica normal.
La evolución fue desfavorable; presentó, desde los 27 días de nacida, convulsiones tónicas con cianosis peribucal tratada con ácido valproico. Sufrió desnutrición grave, síndrome emético agudo con deshidratación y paro cardiorrespiratorio, y falleció a los dos meses.
Caso 3
Lactante menor femenino evaluado por presentar dismorfias faciales (Figura 2). Madre de 27 años de edad, tercera gesta, embarazo simple. Nacimiento por cesárea a las 38 semanas. Requirió de maniobras de estimulación por depresión neonatal moderada. El peso al nacer fue de 2800 g (P 10-25) SDE -0,9 y la talla, de 46 cm (P 3) SDE -2,5. Presentó ictericia neonatal sin complicaciones.

Figura 2. Se aprecia pelo ralo y escaso, hipertricosis en la región frontal, hendiduras palpebrales hacia arriba, puente nasal cóncavo, punta nasal bulbosa y narinas antevertidas, filtrum poco dibujado, labio superior fino e inferior evertido y retrognatia
La resonancia magnética cerebral al mes de edad evidenció ectasia de ventrículos laterales, poligirias focales en el lóbulo parietal izquierdo y quiste subaracnoideo temporal izquierdo.
La sedestación fue a los 21 meses de edad; presentó astigmatismo moderado, paladar hendido, regurgitación y estreñimiento.
Caso 4
Lactante mayor femenino evaluado por presentar retardo global del desarrollo. Madre de 28 años de edad, tercera gesta, embarazo simple, sin complicaciones. Nacimiento por vía vaginal, extrahospitalario, a las 39 semanas. El peso al nacer fue de 3120 g (P 25-50) SDE -0,1 y la talla, de 47 cm (P 3-10) SDE -1,8.
Presentó una convulsión a los siete días de nacida con succión débil, controlada con ácido valproico. La resonancia magnética nuclear reveló un amplio espacio aracnoideo en la región parietal, hidrocefalia e hipoplasia del cuerpo calloso. A los seis meses de edad, se evidenciaron elevaciones en la cifras de triglicéridos y colesterol, además de hipoglucemia e hiperuricemia. La ecografía renal a los nueve meses mostró enfermedad parenquimatosa. Al año de edad, presentó una infección respiratoria; el germen aislado en el cultivo fue Pseudomona aeruginosa, el cual fue sensible a cefotaxima.
Caso 5
Lactante menor masculino evaluado por presentar dismorfias faciales. Primo hermano paterno con trisomía 21. Madre de 33 años de edad, primera gesta, embarazo gemelar bicorial y biamniótico, complicado con hipertensión arterial tratada. Nacimiento por cesárea a las 36 semanas. Fue hospitalizado por los diagnósticos de RCIU, ictericia tratada con fototerapia y persistencia de ducto arterioso de 1,9 mm con cierre exitoso por el tratamiento farmacológico. El peso al nacer fue de 1730 g (P < 3) SDE -3,9 y la talla, de 41 cm (P < 3) SDE -7,3.
La resonancia magnética nuclear cerebral a los cinco meses de edad evidenció retardo de la mielinización, brazos anteriores de los núcleos basales no acorde a la edad en maduración cortical, subcortical y sustancia blanca, ectasia de los ventrículos laterales y adelgazamiento del cuerpo calloso. Presentó criptorquidia izquierda grado I A y pie valgo bilateral congènito.
DISCUSIÓN
Los individuos con la T18 en mosaico presentan un fenotipo muy variable que, en algunos casos, puede dificultar el diagnóstico.4 Ante la presencia de múltiples anomalías congénitas y del desarrollo, se indica realizar el estudio citogenético, el cual permitirá confirmar el diagnóstico y precisar el tipo de alteración cromosómica.5
En ningún caso, se presentó consanguinidad parental y los estudios citogenéticos realizados a los padres fueron normales. En el caso 3, las dismorfias faciales fueron atípicas (Figura 2). Los casos 1 y 2 presentaron fenotipo similar con diferente porcentaje de células con T18. Por su parte, el caso 5, con un menor porcentaje de células con T18 que el caso 4, presentó un fenotipo más grave (Tabla 1). Con esto, se corrobora lo complejo en la correlación clínica. Además, no es posible predecir el porcentaje de células con T18 en las neuronas, gónadas u otros órganos que puede orientar esta correlación.1
La mayor serie de casos estudiados en la literatura incluyó a 33 individuos con T18 en mosaico; la mayoría de los individuos sobrevivieron a largo plazo. Sin embargo, esto no ocurre siempre así. Además, 9/16 (56%) de los casos con menos de 50% de células trisómicas en leucocitos presentaron inteligencia normal. No obstante, 5/16 (31%) de los casos con más de 50% de células trisómicas en leucocitos presentaron inteligencia normal. Con ello, se resalta la dificultad de predecir la supervivencia e inteligencia sobre la base del estudio citogenético.1
La evaluación médica en la T18 debe ser interdisciplinaria con el objeto de brindar oportuna y completa atención médica. En todos los casos, se les brindó apoyo psicológico a los padres. Además, se ha asociado la T18 en mosaico con hepatoblastoma4,6,7 y, en un caso, con linfoma de Hodgkin.2 Es por esto por lo que, aparte de las evaluaciones realizadas, los pacientes deben ser seguidos por la Unidad de Oncología Pediátrica ante el posible riesgo de presentar cáncer.
La realización del estudio citogenético permite ofrecer un oportuno asesoramiento genético; presenta un riesgo de recurrencia menor de 1% en casos de trisomía en mosaico mientras que los padres no sean portadores de alteraciones cromosómicas o la edad materna no aumente este riesgo.
Esta es la mayor serie de casos presentada por una institución, lo que puede deberse a que es un centro de referencia del occidente de Venezuela, ya que los pacientes estudiados son procedentes de cuatro estados diferentes.
1. Tucker ME, Garringer HJ, Weaver DD. Phenotypic spectrum of mosaic trisomy 18: two new patients, a literature review, and counseling issues. Am J Med Genet A 2007;143A(5):505-17. [ Links ]
2. Motta S, Sala D, Sala A, et al. Hodgkin lymphoma in a patient with mosaic trisomy 18: First clinical observation. Am J Med Genet A 2016;170(3):777-80. [ Links ]
3. Banka S, Metcalfe K, Clayton-Smith J.Trisomy 18 mosaicism: report of two cases. World J Pediatr 2013;9(2):179-81. [ Links ]
4. Fernández KS, Baum R, Fung B, et al. Chemoresistant hepatoblastoma in a patient with mosaic trisomy 18 treated with orthotopic liver transplantation. Pediatr Blood Cancer 2011;56(3):498-500. [ Links ]
5. Fitas AL, Paiva M, Cordeiro AI, et al. Mosaic trisomy 18 in a five-month-old infant. Case Rep Pediatr 2013;2013:929861. [ Links ]
6. Pereira EM, Marion R, Ramesh KH, et al. Hepatoblastoma in a mosaic trisomy 18 patient. J Pediatr Hematol Oncol 2012;34(4):e145-8. [ Links ]
7. Ahmad N, Wheeler K, Stewart H, et al. Hepatoblastoma in a mosaic trisomy 18 child with hemihypertrophy. BMJ Case Rep 2016;2016.pii: bcr2015211380. [ Links ]

