










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.115 no.4 Buenos Aires ago. 2017
http://dx.doi.org/10.5546/aap.2017.e220
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2017.e220
Síndrome de Loeys-Dietz, 3 generaciones, 4 casos familiares
Loeys-Dietz Syndrome, 3 generations, 4 familial cases
Dr. Carlos F. Rosentala, Dra. Silvina Neilinga, Dra. Natalia Napolib, Dra. Claudia Villalbaa, Dr. Jorge Barrettab y Dr. Horacio Capellia
a. Hospital General de Pediatría "Prof. Dr. Juan P. Garrahan". Ciudad Autónoma de Buenos Aires.
b. Hospital Italiano de Buenos Aires. Ciudad Autónoma de Buenos Aires.
Correspondencia: Dr. Carlos F. Rosental: cfrosental@yahoo.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 22-9-2016
Aceptado: 21-3-2017
RESUMEN
El síndrome de Loeys-Dietz es una enfermedad genética autosómica dominante caracterizada por aneurismas aórticos, tortuosidad arterial generalizada, hipertelorismo y úvula bifida o paladar hendido. La complicación cardiovascular más grave es la disección aórtica.
Se presentan cuatro casos familiares de este síndrome en tres generaciones, todos con dilatación aórtica grave, y se describen sus aspectos diagnósticos, indicación y tratamiento quirúrgico, como así también pautas de seguimiento.
Palabras clave: Síndrome de Loeys-Dietz; Aneurisma aórtico; Disección aórtica.
ABSTRACT
Loeys-Dietz Syndrome is an autosomal dominant disease with aortic aneurysms, arterial tortuosity with hypertelorism and bifid uvula. We describe four familial cases within three generations. The diagnosis, surgical management and followup will be addressed.
Key words: Loeys-Dietz syndrome; Aortic aneurysm; Aortic dissection.
INTRODUCCIÓN
El síndrome de Loeys-Dietz (Loeys-Dietz Syndrome; LDS, por sus siglas en inglés) es un trastorno del tejido conectivo descrito por primera vez en 2005.1,2 Se trata de una patología de transmisión autosómica dominante caracterizada, principalmente, por presentar aneurismas aórticos, tortuosidad arterial generalizada, hipertelorismo y úvula bífida o paladar hendido. También puede presentar manifestaciones esqueléticas, como escoliosis, pectus carinatum o excavatum y laxitud articular.1-3
El compromiso de la aorta torácica con alto riesgo de aneurismas y posterior disección es la complicación más grave y demanda una actitud intervencionista temprana ante el hallazgo de una dilatación aórtica significativa.4
Afecta tanto a hombres como a mujeres sin distinción étnica. Se desconoce la prevalencia de este síndrome. Es probable que existan pacientes con diagnóstico de alguna otra enfermedad del tejido conectivo que presentan LDS, especialmente, aquellos pacientes con síndrome de Marfan diagnosticados como Marfan "atípico".
Se presentan cuatro casos familiares con LDS en tres generaciones, todos con dilatación aórtica grave, y se mencionan sus aspectos diagnósticos, indicación y tratamiento quirúrgico y pautas de seguimiento.
Presentación del caso
Un niño de 14 años llegó a la consulta cardiológica derivado por su pediatra por presentar un soplo sistólico. Refirió como antecedentes familiares una abuela materna fallecida por disección aórtica.
Al momento del examen físico, se destacaba la presencia de hipertelorismo y, en la auscultación, un soplo sistólico y un clic de eyección sugestivo de válvula aórtica bicúspide. El electrocardiograma era acorde a la edad. En la telerradiografía de tórax, se observaba una silueta cardíaca de tamaño normal, pero con un botón aórtico prominente y flujo pulmonar conservado. En el ecocardiograma doppler color, se constató dilatación significativa de aorta ascendente.
Ante la sospecha de encontrarse frente a un niño con LDS, se revisó la cavidad oral y se detectó la presencia de úvula bífida (Figura 1). La madre refirió que su madre, también operada de una disección aórtica, presentaba la misma úvula característica y la recordaba como "campanita doble".

Figura 1. Úvula bífida
Para optimizar la dimensión y extensión de la dilatación aórtica, se indicó una resonancia magnética nuclear cardíaca. Este estudio demostró que la dilatación aórtica incluía toda la aorta ascendente y su dimensión era 40 mm (Z: + 5),5 sin insuficiencia aórtica. No presentaba otros aneurismas (Figura 2).
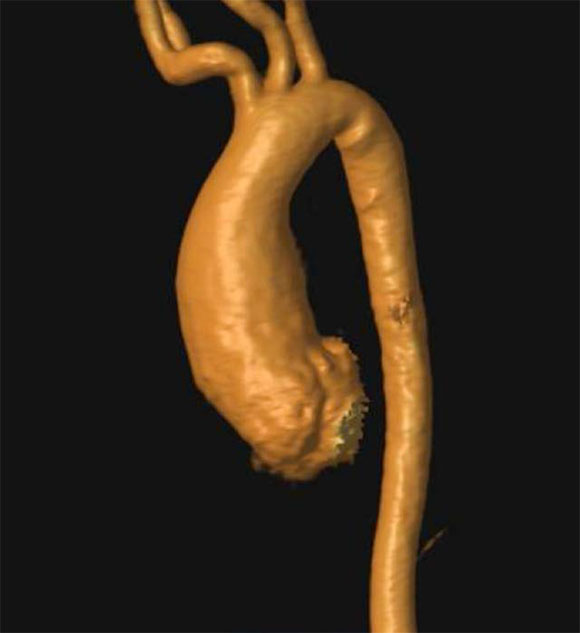
Figura 2. Angiorresonancia magnética nuclear cardíaca. Reconstrucción 3D de aorta
Dado el alto riesgo de disección aórtica, el paciente fue referido a Cirugía Cardiovascular y se realizó una cirugía de Tirone David, consistente en el recambio de toda la aorta ascendente con una prótesis vascular con preservación de la válvula aórtica nativa (Figuras 3 y 4).
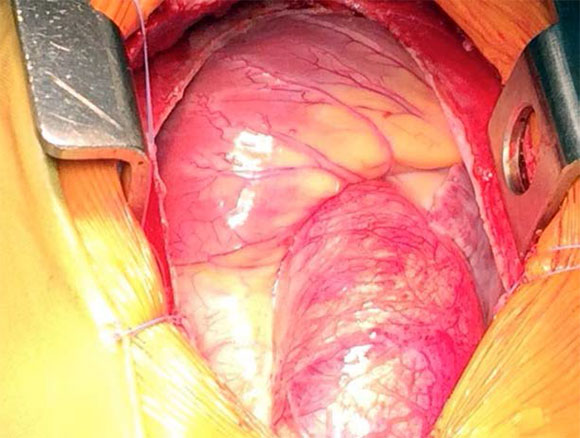
Figura 3. Vista desde la cabecera del paciente, corazón y aorta ascendente dilatada

Figura 4. Vista desde la cabecera del paciente, cirugía de reemplazo de aorta ascendente con preservación de la válvula
A partir de este caso, se realizó una evaluación cardiovascular del grupo familiar cercano. La madre también tenía dilatación de aorta ascendente y úvula bífida y, luego de ser evaluada con los mismos exámenes complementarios, fue intervenida con idéntica técnica quirúrgica que su hijo. Los hermanos del paciente presentaban ecocardiogramas normales.
Ampliando el relevamiento familiar, se detectó que un tío materno presentaba dilatación grave de la raíz aórtica con insuficiencia aórtica moderada, por lo que se indicó cirugía cardiovascular y fue luego intervenido en otro centro con cirugía de Bentall (recambio de la aorta ascendente y reemplazo valvular aórtico con prótesis mecánica).
De este modo, en el término de un mes, gracias a la derivación oportuna del pediatra, un minucioso interrogatorio, un examen físico completo, análisis complementarios y relevamiento familiar, se detectó una patología grave con riesgo de disección aórtica y muerte súbita en tres generaciones y se intervinieron con celeridad y éxito tres integrantes de una misma familia.
DISCUSIÓN
El LDS en una enfermedad genética que se describió, originalmente, por la expresión variable de las mutaciones en el receptor I y II del factor transformador de crecimiento p (transforming growth factor beta receptor 1 and 2; TGFBR1 y TGFBR2, por sus siglas en inglés) y luego se observó en otras mutaciones con manifestaciones típicas de LDS (SMAD3 gen) (TGFB2).3
La enfermedad aneurismática aórtica rápidamente progresiva es una característica distintiva de LDS, lo que requiere una estrecha vigilancia. Los individuos con LDS con alteraciones craneofaciales características (originalmente descrito como LDS I)6 presentan un riesgo muy alto de disección aórtica. En estos casos, la ruptura de los aneurismas puede producirse en edades tempranas y con dimensiones menores que aquellas con otros síndromes de afectación del tejido conectivo, como el Marfan. Se ha publicado disección aórtica en niños de tan solo 3 meses y hemorragia cerebral a partir de los 3 anos7,8 en pacientes con LDS.
Estos pacientes requieren evaluación ecocardiográfica con intervalos frecuentes para monitorear la raíz aórtica y aorta ascendente.
El uso de medicación profiláctica debe ser considerado para los individuos con LDS sin dilatación aórtica si presentan una historia familiar de LDS con dilatación o disección de aorta.9
La dilatación o disección aórtica está presente en el 95% de los casos. Como tratamiento preventivo, se deben restringir los ejercicios competitivos, de contacto o isométricos que aumentan el estrés parietal aórtico y considerar la administración de betabloqueantes,10 bloqueantes de receptor de angiotensina ii y/o inhibidor de la enzima convertidora de angiotensina (IECA).
La enfermedad aneurismática, que incluye la disección, no se limita a la raíz aórtica y se ha informado en todas las porciones de la aorta y ramas de arterias del cuello, aorta torácica y abdominal. Por ello, los estudios de imágenes vasculares de diagnóstico, como la angiografía por resonancia magnética o angiotomografía computarizada con reconstrucción tridimensional de la cabeza, el cuello, el tórax, el abdomen y la pelvis, se deben realizar para evaluar los aneurismas y tortuosidad de la aorta y de todo el árbol arterial.11,12
La decisión de intervención oportuna de la aorta se basa típicamente en la dimensión absoluta de la aorta, la tasa de progresión de la dilatación y la función de la válvula aórtica.13
Dado el mayor riesgo de disección aórtica en pacientes con LDS, la indicación quirúrgica es más precoz. En adultos, se indica con diámetros de raíz aórtica > 4,0 cm (> 4,2 cm según la Sociedad Europea de Cardiología)14 o un crecimiento mayor de 0,5 cm/año. Para niños con anomalías cráneo-faciales graves, originalmente descritas como LDS I, se sugiere la cirugía cuando la raíz aórtica presenta un puntaje Z > +3 o un crecimiento mayor de 0,5 cm/año. Para niños con anomalías cráneo-faciales leves (LDS II), se recomienda la cirugía con puntaje Z de la raíz aórtica > +4 o bien un crecimiento de esta mayor de 0,5 cm/año.
Cuando la insuficiencia valvular aórtica no es significativa, se recomienda la cirugía de reemplazo de raíz aórtica con preservación de la válvula,15 como se indicó y realizó en dos de nuestros 3 casos. La preservación de la válvula tiene la enorme ventaja de evitar la anticoagulación y sus complicaciones, sobre todo, en edad pediátrica. De presentarse disfunción valvular aórtica, la cirugía de Bentall con reemplazo valvular aórtico es el procedimiento de elección, como fue realizado en nuestro tercer paciente.
Dado que la enfermedad aneurismática puede ser progresiva, se debe realizar una evaluación inicial y repetirla con un intervalo de 2 años si no hay aneurismas o disecciones identificados. Así, también en el paciente con LDS operado por dilatación de la raíz aórtica, se recomienda la ecografía posoperatoria con intervalos de 3 a 6 meses durante 1 año después de la cirugía y de 6 meses a 1 año a partir de entonces.6 Otra estrategia complementaria son las medidas para disminuir el estrés parietal (véase Tabla 1).
Tabla 1. Guía para el seguimiento cardiológico y cirugía cardiovascular en pacientes con síndrome de Loeys-Dietz9

Los pacientes pueden necesitar múltiples intervenciones para la aorta y/u otras arterias. Se han informado hasta un 33% de pacientes con intervenciones múltiples.8
Por último, cabe destacar que también se han descrito afectaciones de otros órganos en el LDS que, de presentarse, requieren el abordaje interdisciplinario con especialistas en ortopedia, alergia, gastroenterología, nutrición, neurocirugía, patología vascular periférica, patología espinal, cirugía general, oftalmología, neumonología, psicología para evaluar el impacto psicosocial del diagnóstico en la familia y, por supuesto, la consulta familiar con Genética para decidir el plan de manejo general, consejo genético, chequeo familiar y opciones de estudios durante el embarazo.
En conclusión, el LDS es una enfermedad genética recientemente descrita que presenta similitudes con el síndrome de Marfan, con el que los pediatras están más familiarizados, pero cuya evolución es más desfavorable que este último. El pediatra debe tenerlo presente dentro de sus sospechas diagnósticas por interrogatorio o examen físico y derivar oportunamente al cardiólogo, ya que, en caso de ser confirmado el diagnóstico de dilatación aórtica, presente en el 90% de los casos, obliga a actuar con celeridad debido al alto riesgo de disección aórtica.
1. Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006;355(8):788-98. [ Links ]
2. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37(3):275-81. [ Links ]
3. Van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011;43(2):121-6. [ Links ]
4. Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010;121(13):e266-369. [ Links ]
5. Warren AE, Boyd ML, O'Connell C, et al. Dilatation of the ascending aorta in paediatric patients with bicuspid aortic valve: frequency, rate of progression and risk factors. Heart 2006;92(10):1496-500. [ Links ]
6. Morris SA, Orbach DB, Geva T, et al. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation 2011;124(4):388-96. [ Links ]
7. Malhotra A, Westesson PL. Loeys-Dietz syndrome. Pediatr Radiol 2009;39(9):1015. [ Links ]
8. Williams JA, Loeys BL, Nwakanma LU, et al. Early surgical experience with Loeys-Dietz: a new syndrome of aggressive thoracic aortic aneurysm disease. Ann Thorac Surg 2007;83(2):S757-63; discussion S785-90. [ Links ]
9. MacCArrick G, Black JH 3rd, Bowdin S, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med 2014;16(8):576-87. [ Links ]
10. Gallo EM, Loch DC, Habashi JP, et al. Angiotensin II-dependent TGF-p signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 2014;124(1):448-60. [ Links ]
11. Valverde I, Simpson J, Beerbaum P. Magnetic resonance imaging findings in Loeys-Dietz syndrome. Cardiol Young 2010;20(2):210-3. [ Links ]
12. Morris SA, Orbach DB, Geva T, et al. Increased vertebral artery tortuosity index is associated with adverse outcomes in children and young adults with connective tissue disorders. Circulation 2011;124(4):388-96. [ Links ]
13. Patel ND, Arnaoutakis GJ, George TJ, et al. Valve-sparing aortic root replacement in Loeys-Dietz syndrome. Ann Thorac Surg 2011;92(2):556-60. [ Links ]
14. Erbel R, Aboyans V, Boileau C, et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of aortic diseases of aortic diseases of the European Society of Cardiology (ESC). Eur Heart J 2014;35(41):2873-926. [ Links ]
15. David TE, Feindel CM. An aortic valve-sparing operation for patients with aortic incompetence and aneurysm of the ascending aorta. J Thorac Cardiovasc Surg 1992;103(4):617-21. [ Links ]

