










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.115 no.6 Buenos Aires dic. 2017
http://dx.doi.org/10.5546/aap.2017.e449
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2017.e449
Microdeleción 16p11.2: primeros casos reportados en Argentina
16p11.2 Microdeletion: first report in Argentina
Dra. Agostina Tardivoa, Dra. Bárbara Masottoa, Lic. Lucía Espechea, Dra. Andrea P. Solaria, Dr. Julián Nevadob y Bioq. Sandra Rozentala
a. Centro Nacional de Genética Médica "Dr. Eduardo E. Castilla", Ciudad Autónoma de Buenos Aires.
b. Instituto de Genética Médica y Molecular (INGEMM)/Hospital Universitario La Paz, Instituto de Investigación Sanitaria del Hospital Universitario La Paz (IdiPAZ), Universidad Autónoma de Madrid, Madrid, España.
Correspondencia: Dra. Agostina Tardivo: agostinatardivo@gmail.com
Financiamiento: El trabajo contó con financiación externa. Subsidio de la llI Convocatoria a Fondos Concursables de la Administración Nacional de Laboratorios e Institutos de Salud (ANLIS), año 2011 (FOCANLIS 2011), y una Beca de Investigación "Carrillo Oñativia" de la Comisión Nacional Salud Investiga del Ministerio de salud, año 2012. Ambos, bajo el título "Estudios genómicos en retardo mental idiopático"; directora: Sandra Rozental..
Conflicto de intereses: Ninguno que declarar.
Recibido: 9-3-2017
Aceptado: 27-6-2017
RESUMEN
La microdeleción 16p11.2 se relaciona, habitualmente, con discapacidad intelectual y trastornos del espectro autista. El rango fenotípico incluye un espectro que se extiende desde discapacidad intelectual con o sin autismo, alteraciones del aprendizaje y del lenguaje hasta fenotipos normales. El diagnóstico de la microdeleción se realiza mediante estudios genómicos capaces de identificar variación en número de copias, como la hibridación genómica comparativa en microarreglos, conocida como arrayCGH. Sin embargo, la predicción del fenotipo de un individuo basada únicamente en la localización de dicha deleción sigue siendo un desafío, ya que la existencia de un gran número de variantes en el genoma dificulta la interpretación de posibles efectos funcionales de los genes que contribuyen a dicha región. Se describen dos casos clínicos de pacientes con microdeleción heterocigota en 16p11.2 y se destacan los hallazgos fenotípicos y conductuales que dificultaron la estrategia diagnóstica. También se discuten las implicancias del diagnóstico para el asesoramiento genético familiar.
Palabras clave: Microdeleción 16p11.2; Trastorno del espectro autista; Discapacidad intelectual; ArrayCGH; Herencia oligogénica.
ABSTRACT
The 16p11.2 recurrent microdeletion phenotype is characterized by developmental delay, intellectual disability, and/or autism spectrum disorder. This microdeletion is associated with variable clinical outcome, the phenotypical spectrum ranges from intellectual disability and/or multiple congenital anomalies, autism, learning and speech problems, to a normal phenotype. Genomic testing that determines copy number of sequences, such as chromosomal microarray, is used to identify this microdeletion. However, the prediction of the individual phenotype of a patient based only on the location of such deletion remains a challenge, regarding the existence of many genomic variants that might hinder the interpretation of possible functional effects between most of the contributing genes to that region. We describe the clinical findings in two subjects with heterozygous microdeletions at 16p11.2, highlighting the phenotypic and behavioural findings that conditioned the diagnostic strategy. We also discuss the implications of diagnosis, in practical counselling situations.
Key words: 16p11.2 microdeletion; Autistic spectrum disorder; Intellectual disability; ArrayCGH; Oligogenic inheritance.
INTRODUCCIÓN
La microdeleción 16p11.2 se relaciona, habitualmente, con discapacidad intelectual (DI) y trastornos del espectro autista (TEA). Tiene una prevalencia aproximada de 1/20001 en población general y alcanza el 0, 5-1% en pacientes con TEA.2,3 Se define como la deleción heterocigota recurrente de ~593-kb en la posición aproximada de 29, 6-30, 2 Mb del genoma de referencia (GRCh37/hg19). Si bien, al igual que otros desórdenes genómicos, habitualmente, ocurre de novo, también puede ser transmitida de padres a hijos con patrón autosómico dominante.4
El fenotipo asociado a esta microdeleción es variable. Entre los signos principales, se encuentran macrocefalia y TEA.5-7 Además, incluye predisposición a una forma de obesidad de alta penetrancia, que incrementa 43 veces el riesgo de desarrollar obesidad mórbida.8 Se ha descrito un "fenómeno en espejo" asociado a microduplicaciones de esta región, que se presentan con bajo peso, microcefalia y esquizofrenia.9,10
El diagnóstico se realiza mediante estudios genómicos capaces de identificar variación en el número de copias (copy number variation; CNV, por sus siglas en inglés), como la hibridación genómica comparativa en microarreglos (array comparative genomic hybridization; arrayCGH, por sus siglas en inglés). No puede detectarse en el cariotipo con bandeo G ni otras técnicas de bandeo de la citogenética convencional.
El objetivo de este trabajo es presentar los hallazgos clínicos y moleculares en dos pacientes con microdeleción 16p11.2 y destacar los hallazgos fenotípicos y conductuales que dificultaron la estrategia diagnóstica.
PRESENTACIÓN DE CASOS CLÍNICOS
Se analizaron las historias clínicas de dos pacientes con diagnóstico de microdeleción 16p11.2 de nuestro Centro. En ambos pacientes, se realizaron los siguientes estudios genéticos: estudio citogenético con bandeo G, 25 metafases, 550 bandas. Estudio del gen FMR1: reacción en cadena de la polimerasa (polymerase chain reaction; PCR, por sus siglas en inglés) y Southern de pacientes evaluados para cada ítem. NR: no reportado. p: número de pacientes. SNC: sistema nervioso central.
Se menciona SÍ o NO según se haya descrito o no la anomalía; en caso de encontrarse, se explicita el número de casos sobre el total de pacientes evaluados para cada ítem. NR: no reportado. p: número de pacientes.
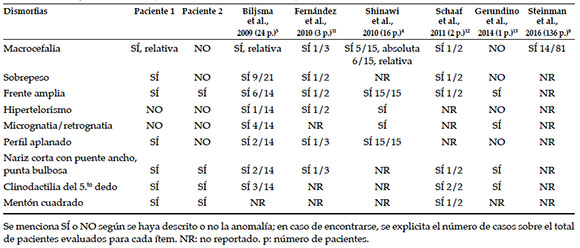
blot. Amplificación de sondas dependiente de ligandos múltiples (multiplex ligation dependent probe amplification; MLPA, por sus siglas en inglés) para deleciones subteloméricas: kits comerciales SALSA MLPA P036-E2 Telomere-3 y SALSA MLPA P070-B2 Human Telomere-5 (MRC-Holland, The Netherlands). ArrayCGH: array a medida de 8x60K (Karyoarray® basado en la plataforma Agilent SurePrint G3 CGH), desarrollado por el Instituto de Genética Médica y Molecular (INGEMM), y el diseño SurePrint G3 CGH ISCA v2 8x60K de Agilent. Se compararon los hallazgos fenotípicos con los reportes y series descritos en la literatura (anomalías asociadas y dismorfias, Tablas 1 y 2).2,4-6,8,11-13
Tabla 1. Anomalías asociadas descritas en pacientes con microdeleción 16p11.2
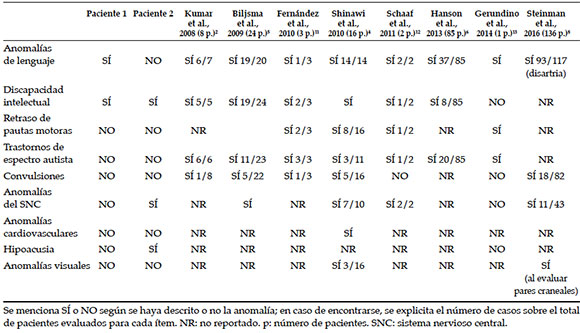
Tabla 2. Dismorfias descritas en pacientes con microdeleción 16p11.2
CASOS CLÍNICOS
Paciente 1 (Figura 1.A):
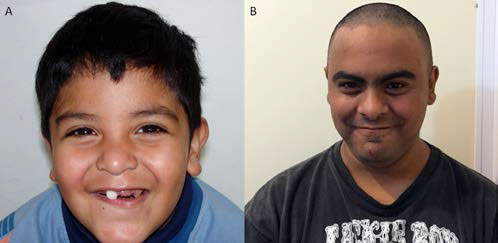
Figura 1. Pacientes con microdeleción 16p11.2 estudiados por nuestro Centro. A: Paciente 1. B: Paciente 2
Varón de 4 años que consultó por DI, con trastornos de lenguaje e hiperactividad sin retraso motor. Se desconocían antecedentes familiares paternos; un tío y una prima tenían DI por vía materna. No presentaba antecedentes perinatales de relevancia. Recién nacido de término (RNT) con peso adecuado para la edad gestacional (PAEG) de 3120 g. Puntaje de Apgar referido vigoroso.
Al momento del examen físico, a los 6 años, presentaba perímetro cefálico (PC): 52 cm en percentilo (Pc) + 1 DE; peso: 26 kg en Pc 7590; talla: 114 cm en Pc 25, macrocefalia relativa, frente hirsuta, nariz de dorso recto, puente ancho y punta redonda, hipodontia, barbilla marcada y prominente, orejas displásicas y desplegadas, hiperlaxitud metacarpofalángica y hallux ancho bilateral.
Los estudios complementarios, audiometría, fondo de ojo (FO), ecografía abdominorrenal, ecocardiograma, electroencefalograma, potenciales evocados auditivos, tomografía axial computada y resonancia nuclear magnética (RNM) cerebrales, resultaron normales. El coeficiente intelectual (CI), evaluado en dos oportunidades, fue de 79 (2009) y de 58 (2011).
Resultados de los estudios genéticos. Cariotipo: 46, XY [30]. PCR gen FMR1: 30 repeticiones. Southern blot gen FMR1: banda de 2, 8kb (normal). MLPA para deleciones subteloméricas: rsa(P036, P070)x2 (normal). ArrayCGH: arr[hg19]1p36.33 (16278991663760) x1, 1q42.13 (22783291827926392) x3, 16p11.2 (16: 29674336-30199351) x1. Deleción 16p11.2: CNV patogénica. Deleción 1p36.33: CNV benigna. Duplicación 1q42.13: variante de significado incierto (variant of unknown significance; VOUS, por sus siglas en inglés), sin evidencia de patogenicidad ni asociación con el fenotipo a la fecha. Se realizó arrayCGH a su madre: arr[hg19](1-22, X)x2 (Normal). Su padre no estaba disponible para el estudio.
Paciente 2 (Figura 1.B):
Varón de 17 años que consultó por retraso madurativo y facies peculiar, sin retraso de pautas motoras.
Antecedentes familiares: madre con lupus eritematoso sistémico y dificultad para finalizar la escuela primaria. Antecedentes perinatológicos: madre medicada con fenobarbital durante el primer trimestre del embarazo. RNT/PAEG (3620 g). Puntaje de Apgar referido vigoroso. Pie bot.
Al momento del examen físico, presentaba PC: 56 cm en Pc 50; peso: 60 kg en Pc 25-50; talla: 163 cm en Pc 25, con implantación anterior del cabello alta con pico de viuda, frente amplia, cejas pobladas con sinofris; nariz de dorso recto con columela prominente y punta redonda, filtrum liso, ligero prognatismo y paladar alto, barbilla marcada y prominente, orejas displásicas con hélix grueso, manos con clinodactilia bilateral del 5.to dedo e hiperlaxitud metacarpofalángica, hallux ancho bilateral y micropene.
En los estudios complementarios, se detectó hipoacusia neurosensorial leve y, en la RNM cerebral, un quiste aracnoideo centro-axial en fosa posterior.
Resultados de los estudios genéticos. Cariotipo: 46, XY [20]. PCR gen FMR1: 31 repeticiones (normal). MLPA para deleciones subteloméricas: rsa11p15.5 (P036, P070) x3mat: duplicación heterocigota en 11p15.5. ArrayCGH: arr[hg19]11p15.5(196966-215068) x3, 16p11.2(29674336-30198123)x1~2 (Figura 2). Deleción 16p11.2: CNV patogénica. Duplicación 11p15.5: CNV benigna. El estudio de arrayCGH de ambos padres se encuentra pendiente.
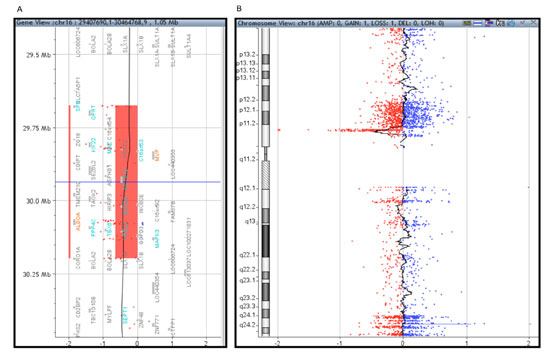
Figura 2. Visualización de la deleción en 16p11.2 con el programa Cytogenomics (Agilent) para ambos pacientes. A. Visualización de la deleción y los genes involucrados. B. Visualización de la deleción en el contexto del cromosoma
DISCUSIÓN
La microdeleción 16p11.2 (OMIM # 611913), que se considera un locus de susceptibilidad para autismo (AUTS14A), fue descrita por Barnby y cois., en 20059 como síndrome de deleción 16p11.2.
La región contiene, aproximadamente, 25 genes y, al menos, 17 de ellos se expresan en el cerebro de mamíferos y juegan un rol importante en el neurodesarrollo. Entre estos genes, MAPK3, DOC2A y SEZ6l2 estarían asociados a los trastornos conductuales.10 MAPK3 (MIM 601795) codifica para una proteína de señalización sináptica que interviene en los procesos neurológicos del aprendizaje y DOC2A (MIM 604567) codifica para una proteína que se expresa, predominantemente, en el cerebro y estaría implicado en la liberación de neurotransmisores dependientes de calcio. KCTD13 estaría asociado a la microcefalia, aunque MAPK3 y MVP serían posibles modificadores de este proceso.10 A medida que aumente el conocimiento de este síndrome, se podrá establecer con mayor precisión el rol de estos genes en el fenotipo.
En nuestros pacientes, existen características coincidentes en el aspecto clínico y conductual con lo descrito en varios de estos reportes, como DI, TEA y trastornos del lenguaje (el primero de ellos es el hallazgo más constante). El sobrepeso, presente en el primer caso, se trata de un hallazgo comunicado, aproximadamente, en la mitad de los pacientes en las series analizadas.6,11,14 Además, la presencia de convulsiones no se observó en ninguno de nuestros pacientes.4,5,8 La hipoacusia neurosensorial sin historia familiar ni personal que justifique otra etiología, presente en el paciente 2, no es un hallazgo comunicado previamente en portadores de la deleción.
Si bien algunas de las dismorfias resultan concordantes, estas no permiten por sí solas orientar el diagnóstico. La derivación oportuna al Servicio de Genética es fundamental en estos pacientes. En los casos con confirmación de síndrome de deleción 16p11.2, se recomienda el seguimiento por Genética, Neurología, Psicopatología, Otorrinolaringología, Cardiología, Traumatología y Nutrición (en el caso de detectar sobrepeso).
El asesoramiento genético presenta una dificultad: se trata de una microdeleción que se encuentra también en individuos sanos, por lo que se incluye esta anomalía dentro de la llamada herencia oligogénica, en la que otros factores, ambientales o genes modificadores, dan manifestación clínica variable desde individuos sanos hasta afectados con DI.15 A la fecha, se sugiere un patrón dominante con penetrancia incompleta, por lo que no puede estimarse de forma certera el riesgo de ocurrencia de anomalías fenotípicas en la descendencia de portadores sanos. Se espera que estudios futuros en grupos familiares permitan dar una mejor respuesta a los mecanismos involucrados. La microdeleción detectada en el paciente 2 se encontraría en mosaico. Esta afección podría contribuir a la penetrancia incompleta y expresividad variable observada. La descripción fenotípica de otros casos similares contribuirá a clarificar este punto.4
La prevalencia basada en los reportes internacionales indica que esta anomalía está subdiagnosticada en nuestra población. Esto estaría relacionado con el acceso limitado a los estudios de arrayCGH y con el asesoramiento genético en la Argentina. Considerando que diferentes comunicaciones y consensos internacionales recomiendan el arrayCGH como el primer paso en el protocolo de diagnóstico genético en pacientes con DI, TEA y anomalías congénitas múltiples,14 es importante empezar a integrar esta herramienta en genética clínica. Además del incremento en la sensibilidad y especificidad, el arrayCGH reduce la complejidad del diseño de estrategias o protocolos para establecer el diagnóstico de certeza en pacientes con DI, por lo que es de especial relevancia cuando estos pacientes no presentan un fenotipo orientador.
La comunicación de los casos presentados aporta nuevas evidencias en la descripción del síndrome de microdeleción 16p11.2. Al igual que otros desbalances genómicos, se trata de una entidad poco frecuente y, por lo tanto, es necesaria la contribución de muchos grupos de trabajo para su caracterización.
1. Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011;478(7367): 97-102. [ Links ]
2. Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008;17(4): 628-38. [ Links ]
3. Miller DT, Chung W, et al. 16p11.2 Recurrent Microdeletion. In: Pagon RA, Adam MP, Ardinger HH, et a., editors. Gene Reviews®. Seattle (WA): University of Washington; 2009.
4. Shinawi M, Liu P, Kang SHL, et al. Recurrent reciprocal 16p11.2rearrangementsassociatedwithglobaldevelopmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. JMed Genet 2009;47(5): 332-41. [ Links ]
5. Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet 2009; 52(2-3): 77-87. [ Links ]
6. Hanson E, Bernier R, Porche K, et al. The Cognitive and Behavioral Phenotype of the 16p11.2 Deletion in a Clinically Ascertained Population. Biol Psychiatry 2015;77(9): 785-93. [ Links ]
7. Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 2010;463(7281): 671-5. [ Links ]
8. Steinman KJ, Spence SJ, Ramocki MB, et al. 16p11.2 deletion and duplication: Characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A 2016;170(11): 2943-55. [ Links ]
9. Barnby G, Abbott A, Sykes N, et al. Candidate-gene screening and association analysis at the autism-susceptibility locus on chromosome 16p: evidence of association at GRIN2A and ABAT. Am J Hum Genet 2005;76(6): 950-66. [ Links ]
10. Chang H, Li L, Li M, et al. Rare and common variants at 16p11.2 are associated with schizophrenia. Schizophr Res 2017;184: 105-8. [ Links ]
11. Fernandez BA, Roberts W, Chung B, et al. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet 2010;47(3): 195-203. [ Links ]
12. Schaaf CP, Goin-Kochel RP, Nowell KP, et al. Expanding the clinical spectrum of the 16p11.2 chromosomal rearrangements: three patients with syringomyelia. Eur J Hum Genet 2011;19(2): 152-6. [ Links ]
13. Gerundino F, Marseglia G, Pescucci C, et al. 16p11.2 de novo microdeletion encompassing SRCAP gene in a patient with speech impairment, global developmental delay and behavioural problems. Eur JMed Genet 2014; 57(11-12): 649-53. [ Links ]
14. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86(5): 749-64. [ Links ]
15. De Wolf V, Brison N, Devriendt K, et al. Genetic counseling for susceptibility loci and neurodevelopmental disorders: the del15q11.2 as an example. Am J Med Genet A 2013;161A(11): 2846-54. [ Links ]

