










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.1 Buenos Aires Feb. 2018
http://dx.doi.org/10.5546/aap.2018.e130
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2018.e130
Incisivo central único de la línea media del maxilar, holoprosencefalia y estenosis congènita del orificio nasal anterior en un recién nacido prematuro: a propósito de un caso
Dr. Ozkan Ilhana, Prof. Asoc. Dra. Yeliz Pekcevikb, Dra. Sinem Akbayc, Dra. Senem A. Ozdemird, Dra. Seyma Memurc, Dr. Berat Kanarc, Dr. Ozgur Kirbiyik y Prof. Dra. Esra A. Ozerc
a. Facultad de Medicina de Harran Üniversitesi, Departamento de Neonatología, Sanliurfa, Turquía.
b. Hospital Universitario y de Investigación de Tepecik, Departamento de Radiología, Esmirna, Turquía.
c. Hospital Universitario y de Investigación de Tepecik, Departamento de Neonatología, Esmirna, Turquía.
d. Hospital Pediátrico Dr. Behget Uz, Departamento de Neonatología, Esmirna, Turquía.
e. Hospital Universitario y de Investigación de Tepecik, Centro de Diagnóstico Genético, Esmirna, Turquía.
Correspondencia: Dr. Ozkan Ilhan: ozkanilhan-83@hotmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 19-5-2017
Aceptado: 9-8-2017
RESUMEN
El síndrome del incisivo central único de la línea media del maxilar es un trastorno raro que implica anomalías de la línea media, como holoprosencefalia, anomalías de las fosas nasales, fisura palatina, labio leporino, hipotelorismo, microcefalia y panhipopituitarismo. La estenosis congénita del orificio nasal anterior es una causa mortal de dificultad respiratoria neonatal debido al estrechamiento del orificio nasal anterior, y podría confundirse con la atresia de coanas. En este informe, presentamos el caso de un recién nacido con síndrome del incisivo central único de la línea media del maxilar acompañado de otras anomalías, tales como holoprosencefalia, estenosis del orificio nasal anterior, microcefalia y panhipopituitarismo. El cariotipado mostró una deleción heterocigota en el gen SIX3 en la región 2p21, que produjo una forma más grave de holoprosencefalia.
Palabras clave: Estenosis congénita del orificio nasal anterior; Incisivo central único de la línea media del maxilar; Holoprosencefalia; Hipopituitarismo; Dificultad respiratoria.
INTRODUCCIÓN
El incisivo central único de la línea media del maxilar (SMMCI, por sus siglas en inglés) es una afección rara que puede presentarse como una anomalía dental aislada o como parte del síndrome del SMMCI. Es un trastorno autosómico dominante complejo; la incidencia es de 1/50 000 nacidos vivos.1,2 Este síndrome puede estar acompañado de defectos en la línea media, tales como holoprosencefalia, anomalías de las fosas nasales, fisura palatina, labio leporino, hipotelorismo, microcefalia y panhipopituitarismo. Se observan parto prematuro, bajo peso al nacer y restricción del crecimiento.3,4
En este artículo, presentamos el caso de un recién nacido con síndrome del SMMCI acompañado de otras anomalías, tales como holoprosencefalia, estenosis del orificio nasal anterior, microcefalia y panhipopituitarismo.
A propósito de un caso
Esta paciente nació a las 32 semanas de gestación por parto vaginal espontáneo de una madre sana de 30 años de edad que cursaba su cuarto embarazo y de un padre de 35 años de edad sin consanguinidad con la madre; su peso al nacer fue de 1800 gramos. Según el relato de los padres, a la bebé se la intubó y se la conectó a un respirador en un hospital privado debido a atresia de coanas y panhipopituitarismo. Una vez que trasladaron a la paciente a nuestro centro a los 64 días de vida para que le realicen otras pruebas y continuar con el tratamiento, se la ingresó a la unidad de cuidados intensivos neonatales. Una vez ingresada, su estado general era deficiente, su presión arterial era de 79/45 (56) mmHg, su frecuencia cardíaca era de 130/min, su temperatura corporal era de 36,4 °C, su peso corporal era de 2180 gramos (< 3 p), su talla era de 44 cm (< 3 p) y su perímetro cefálico era de 28 cm (< 3 p). La paciente tenía hipotonía y microcefalia, carecía del frenillo del labio y presentaba paladar en forma de V e hipotelorismo. Se le colocó una sonda nasogástrica de calibre 5F a través de ambas fosas nasales. La paciente había recibido hidrocortisona, levotiroxina y desmopresina en el centro que la derivó; según los análisis de laboratorio, el hemograma completo y la concentración de glucosa eran normales y no se observaron hallazgos anatomopatológicos en el análisis bioquímico además de hipernatremia (154 mmol/l). Las pruebas funcionales de tiroides estaban dentro de los límites normales. La gasometría y la radiografía de tórax eran normales. Se aumentó la dosis de desmopresina según las recomendaciones de un endocrinólogo pediátrico. En la repetición de los análisis, la concentración de sodio y la densidad urinaria para detectar una posible diabetes insípida fueron normales. Se redujo gradualmente el uso de la oxigenoterapia y el respirador durante el seguimiento; se interrumpió el uso de la oxigenoterapia.
Se realizó una tomografía computarizada (TC) craneofacial. Las secuencias axiales de TC al nivel del orificio nasal anterior mostraron prominencias nasales desplazadas de la línea media al nivel del maxilar y estrechamiento del orificio nasal anterior. En los niveles inferiores, se observó un incisivo único resultado de la fusión de los incisivos centrales al nivel de los incisivos anteriores (Figura 1). La secuencia coronal ponderada en T1 de la resonancia magnética nuclear (RMN) craneofacial mostró un ventrículo cerebral único que coincide con holoprosencefalia semilobular. En la imagen tridimensional reconstruida mediante secuencias ponderadas en T1 se observó hipotelorismo y estrechamiento del orificio nasal anterior (Figura 2). Sobre la base de los hallazgos de la TC y la RMN craneofaciales, a la paciente se le diagnosticó síndrome del SMMCI acompañado de estenosis del orificio nasal anterior congénita y holoprosencefalia semilobular.
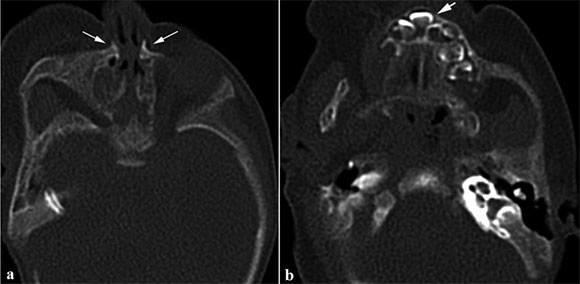
Figura 1. Secuencias axiales de TC al nivel del orificio nasal anterior que muestran prominencias nasales desplazadas de la línea media al nivel del maxilar (flechas) y estrechamiento del orificio nasal anterior. El diámetro del orificio nasal anterior era de 3 mm (a). En el nivel inferior, se observa un incisivo único (flecha) resultado de la fusión de los incisivos centrales al nivel de los incisivos anteriores. Se observa un estrechamiento en forma de V en la bóveda del paladar y prominencias óseas en la línea media (b)

Figura 2. Secuencia coronal ponderada en T1 de la RMN craneofacial en la que se muestra un ventrículo cerebral único que coincide con holoprosencefalia semilobular (a). Imagen tridimensional reconstruida mediante secuencias ponderadas en T1 que muestra el estrechamiento del orificio nasal anterior (flecha) e hipotelorismo (b)
En los exámenes oftalmológicos no se encontraron hallazgos anatomopatológicos. El ecocardiograma fue normal. El cariotipado mostró una deleción heterocigota en el gen SIX3 en la región 2p21.
Se consultó a un otorrinolaringólogo y a un neurocirujano, y no se consideró la realización de una cirugía. Se administraron descongestivos nasales y gotas nasales con dexametasona con fines conservadores durante 15 días.
Se evaluó a la paciente para detectar anomalías futu ras que pudieran aparecer durante la dentición y se la derivó a un ortodoncista. Dado que la paciente no podía alimentarse por boca, se capacitó a los padres para que usaran una sonda orogástrica y se indicó el alta de la paciente a los 81 días de vida. La paciente continúa en seguimiento con un equipo multidisciplinario en nuestro hospital.
DISCUSIÓN
El síndrome del SMMCI es una anomalía rara del desarrollo. Puede ocurrir como una anomalía dental aislada pero también puede estar acompañado de varios defectos de la línea media. Scott5 fue el primero en describirlo en 1958; la etiología de este síndrome está asociada con la mutación en el gen Sonic Hedgehog (SHH) en el cromosoma 7q36.3.4
El síndrome del SMMCI se explica por la fusión de dos dientes de leche y los incisivos centrales permanentes del maxilar, lo que da como resultado un diente único, y esto ocurre entre las 35 y las 38 semanas de gestación. El síndrome del SMMCI puede estar acompañado de defectos de la línea media en las fosas nasales, holoprosencefalia, microcefalia, fisura palatina, labio leporino, anomalías palatinas, hipotelorismo, anomalías de la silla turca, anomalías de la hipófisis y cardiopatías congénitas. Pueden observarse parto prematuro, bajo peso al nacer y restricción del crecimiento, como en el caso de nuestra paciente.2,4,6
La holoprosencefalia es la afección más frecuente en compañía del síndrome del SMMCI. La holoprosencefalia es un defecto del desarrollo que se produce por la separación incompleta del prosencéfalo durante el período embrionario, lo que afecta el desarrollo de las estructuras de la línea media anterior. Los hallazgos en los pacientes con holoprosencefalia incluyen microcefalia, retraso mental, hipotelorismo ocular, fisura palatina, labio leporino e incisivo único de la línea media del maxilar.7 Se considera que una mutación de aminoácido en el gen SHH del cromosoma 7 y una deleción en el gen TGIF del cromosoma 18 son responsables de la holoprosencefalia.2 En algunos casos se han notificado mutaciones en los genes SIX3 y ZIC2.8 Se detectó una deleción heterocigota en el gen SIX3 en la región 2p21 durante el cariotipado de esta paciente. En la bibliografía, se notificaron más de 100 casos con mutación en el gen SIX3 que produjo holoprosencefalia. El gen SIX3 es el tercero entre los genes causantes de holoprosencefalia. En comparación con otras mutaciones, la mutación en el gen SIX3 provoca una forma más grave de holoprosencefalia, aunque aún no se explica el motivo de ello.9
Los recién nacidos respiran solamente a través de sus fosas nasales durante los primeros meses de vida. Los pacientes con síndrome del SMMCI pueden presentar dificultad respiratoria grave debido a la obstrucción nasal durante los primeros días de vida. Esto puede ser debido a la atresia de coanas, la estenosis medionasal o el estrechamiento del orificio nasal anterior.10
El orificio nasal anterior es la porción más estrecha de las vías nasales y una reducción de su diámetro produce dificultad respiratoria al aumentar la resistencia de las vías. Si bien aún no se comprenden cabalmente el desarrollo y la etiología del estrechamiento del orificio nasal anterior, su causa es la excrecencia de los huesos nasales y maxilares. En un recién nacido con dificultad respiratoria, debe considerarse el estrechamiento del orificio nasal anterior si se observa dificultad durante la intubación o colocación de una sonda nasogástrica. El diagnóstico se confirma mediante TC y el criterio diagnóstico es un ancho del orificio nasal anterior inferior a 11 mm. Esta paciente tenía estrechamiento del orificio nasal anterior pero no presentaba atresia de coanas. En las secuencias de TC se observaron prominencias nasales desplazadas de la línea media al nivel del maxilar y estrechamiento del orificio nasal anterior, cuyo diámetro era de 3 mm.10 En una serie de 20 pacientes con estrechamiento del orificio nasal anterior informada por Van Den Abbeele y col.11, se halló síndrome del SMMCI en el 60% de los pacientes.
Entre los pacientes con síndrome del SMMCI, del 10% al 50% podrían presentar un trastorno morfológico acompañante en la silla turca y la hipófisis.2 No fue posible visualizar claramente la hipófisis en la RMN del cráneo de esta paciente; sin embargo, se observaron síntomas de panhipopituitarismo. De manera similar, se observa hipotelorismo leve en el 45% de los pacientes con este síndrome.2 En las imágenes de la RMN de esta paciente se halló hipotelorismo. Otras manifestaciones y anomalías acompañantes fueron parto prematuro, restricción del crecimiento y microcefalia.
Las opciones de tratamiento incluyen métodos conservadores y quirúrgicos según la gravedad de los síntomas. Van Den Abbeele y col.11 recomendaron el uso de gotas nasales con corticoesteroides y epinefrina como manejo inicial y limitaron la duración de este tratamiento a 15 días debido a los efectos secundarios. Lee y col.,12 informaron la realización de cirugías en los pacientes con apnea del sueño, intentos de intubación repetidos o imposibilidad de extubación, problemas alimentarios acompañados de cianosis y pacientes que no respondieron al tratamiento conservador. La intervención quirúrgica más ampliamente recomendada es la incisión sublabial mediante la cual se elimina la excrecencia para incrementar el diámetro del orificio nasal anterior a más de 11 mm. Después de la cirugía, en la mayoría de los casos se inserta una cánula nasal para conservar la permeabilidad de la vía respiratoria y evitar la reestenosis, y la cánula se retira entre 1 y 3 semanas después.12 No se consideró realizar una intervención quirúrgica en esta paciente debido a que no tenía apnea, a que no requirió asistencia respiratoria durante el seguimiento y a que no presentaba cianosis durante la alimentación. Se administraron descongestivos nasales y gotas nasales con dexametasona con fines conservadores durante 15 días.
A modo de conclusión, deben considerarse las enfermedades de las fosas nasales, como la atresia de coanas, la estenosis medionasal o el estrechamiento del orificio nasal anterior, en los recién nacidos con anomalías de la línea media y dificultad respiratoria, y deben guiarse las pruebas diagnósticas y el tratamiento para detectar estas afecciones en forma oportuna.
1. Johnson N, Windrim R, Chong K, et al. Prenatal diagnosis of solitary median maxillary central incisor syndrome by magnetic resonance imaging. Ultrasound Obstet Gynecol 2008;32(1):120-2. [ Links ]
2. Hall RK. Solitary median maxillary central incisor syndrome. Orphanet J Rare Dis 2006;1:12. [ Links ]
3. Barcelos R, Nivoloni Tannure P, Farinhas JA, et al. Solitary median maxillary central incisor in two healthy siblings: case report. J Disabil Oral Health 2011;12(3):133-5. [ Links ]
4. Stone DL, McKusick VA. Solitary median maxillary central incisor; SMMCI. OMIM# 147250. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. 2004 [Acceso: 11 de noviembre de 2016] Disponible en: https://www.omim.org/entry/147250?search=smmci%20 syndrome&highlight=syndromic%20smmci%20 syndrome>.
5. Scott DC. Absence of upper central incisors. Br Dent J 1958;104:247. [ Links ]
6. Machado E, Machado P, Grehs B, et al. Solitary median maxillary central incisor syndrome: case report. Dental Press J Orthod 2010;15(4):55-61. [ Links ]
7. Erdil A, Ertekin V, Alp H, et al. Holoprozensefali: olgu sunumu. Tip Araştirmalari Dergisi 2014;12(2):77-9.
8. Johnson N, Windrim R, Chong K, et al. Prenatal diagnosis of solitary median maxillary central incisor syndrome by magnetic resonance imaging. Ultrasound Obstet Gynecol 2008;32(1):120-2. [ Links ]
9. Lacbawan F, Solomon BD, Roessler E, et al. Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J Med Genet 2009;46(6):389-98. [ Links ]
10. Blackmore K, Wynne DM. A case of solitary median maxillary central incisor syndrome with bilateral pyriform aperture stenosis and choanal atresia. Int J Pediatr Otorhinolaryngol 2010;74(8):967-9. [ Links ]
11. Van Den Abbeele T, T riglia JM, Francois M, et al. Congenital nasal pyriform aperture stenosis: diagnosis and management of 20 cases. Ann Otol Rhinol Laryngol 2001;110(1):70-5. [ Links ]
12. Lee KS, Yang CC, Huang JK, et al. Congenital pyriform aperture stenosis: surgery and evaluation with threedimensional computed tomography. Laryngoscope 2002;112(5):918-21. [ Links ]

