










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.3 Buenos Aires June 2018
¿CUÁL ES SU DIAGNÓSTICO?
Resolución del caso presentado en el número anterior
Dermatitis en un lactante, no siempre benigna
Dermatitis in an infant, not always benign
Dra. Natalia Bejarano Ramíreza, Dra. Julia Pareja Grandea, Dra. Pamela Zamberk Majlisb, Dra. Pilar Cortina de la Calleb y Dr. Francisco J. Redondo Calvoc
a. Servicio de Pediatría, Hospital General Universitario de Ciudad Real, Ciudad Real, España.
b. Servicio de Dermatología, Hospital General Universitario de Ciudad Real, Ciudad Real, España.
c. Servicio de Anestesiología y Reanimación, Hospital General Universitario de Ciudad Real, Ciudad Real, España.
Correspondencia: Dra. Natalia Bejarano Ramírez: nbejarano@sescam.jccm.es
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 21-3-2017
Aceptado: 22-8-2017
Histiocitosis de células de Langerhans Caso clínico
Un niño de 6 meses, con antecedentes personales de prematuro tardío de bajo peso para la edad gestacional (gemelar), consulta por la aparición de lesiones papulares rosado-amarillentas descamativas en el tronco, la espalda y la cara desde hace 1 mes (Figura 1).
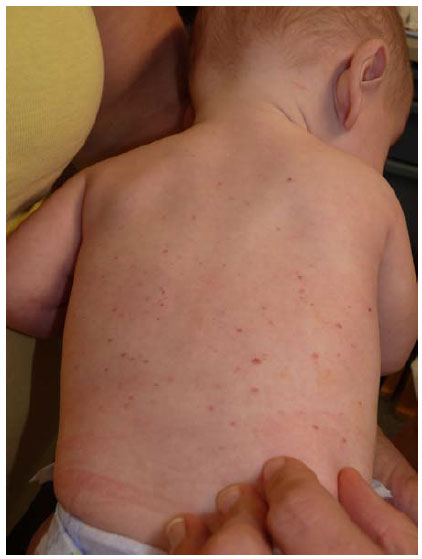
Figura 1. Lesiones en la espalda a los 6 meses de edad
En los últimos 10 días, se aprecia extensión de dichas lesiones hacia las axilas, las ingles y la región anogenital. La familia no refiere fiebre acompañante ni otra clínica asociada. En la exploración, no se observan megalias abdominales.
Se realiza una biopsia cutánea, en la que se observan células largas ovoideas de bordes mal definidos, con citoplasma eosinófilo y núcleo excéntrico en forma de grano de café. Marcadores inmunohistoquímicos positivos para S100 y CD1a. Con microscopio electrónico, se observan gránulos de Birbeck en la mayoría de las células. El infiltrado se localiza en la epidermis y la dermis adventicial.
Con el diagnóstico de histiocitosis de células de Langerhans, se realiza hemograma, función hepática, coagulación, orina, radiografía de tórax, ecografía abdominal y serie ósea, que son normales. Se procede al tratamiento conservador con corticoides tópicos en las lesiones cutáneas y seguimiento del paciente.
A los 12 meses de vida, presenta empeoramiento de las lesiones cutáneas (Figura 2), con afectación hematológica (anemia y trombopenia) y hepática. Se inicia un tratamiento citostático de 1a y 2a línea para histiocitosis, sin respuesta, y precisa trasplante alogénico de médula ósea del hermano gemelo. Evolución favorable, con desaparición de las lesiones cutáneas.
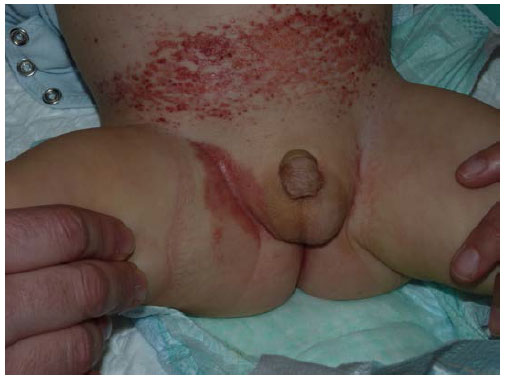
Figura 2. Lesiones en la región inguinal a los 12 meses de edad
Histiocitosis de células de Langerhans
La incidencia de la histiocitosis de células de Langerhans se estima en 5 casos/1 000 000 de habitantes al año. La etiopatogenia es desconocida, aunque se postula una alteración en la regulación del sistema inmunológico de los pacientes afectos, que conlleva la acumulación de macrófagos, células dendríticas y células derivadas de monocitos en diversos órganos y tejidos. Desde la primera clasificación en 1987, se han identificado una serie de nuevos hallazgos en cuanto al origen celular, la patología molecular y las características clínicas de los trastornos histiocíticos. Según los últimos artículos de revisión, se establece una nueva clasificación de histiocitosis sobre la base de histología, fenotipo, alteraciones moleculares y características clínicas y de imagen. Se describen cinco grupos de enfermedades: grupo L (histiocitosis relacionadas con Langerhans), grupo C (histiocitosis cutáneas y mucocutáneas), grupo M (histiocitosis malignas), grupo R (enfermedad de Rosai-Dorfman) y grupo H (linfohistiocitosis hemofagocítica y síndrome de activación de macrófagos).1
Las manifestaciones cutáneas son muy frecuentes y, en más del 35-40% de los casos, suelen ser el primer signo de la enfermedad. Se caracterizan por pápulas rosado-amarillentas descamativas en áreas seborreicas del tronco, la cara y el cuero cabelludo, aunque también pueden observarse erosiones crónicas en la región retroauricular, las axilas, las ingles y la región anogenital. La distribución "en camiseta" de las lesiones, como en nuestro paciente, se ha descrito como patognomónica. En niños pequeños, el diagnóstico diferencial incluye la dermatitis atópica/eczema, escabiosis y otros exantemas vesiculoampollosos, como la varicela. El diagnóstico se realiza sobre la base de la sospecha clínica y se confirma por biopsia. Histopatológicamente, se describe una proliferación de células dendríticas presentadoras de antígenos con características fenotípicas y ultraestructurales específicas. Las células de Langerhans son células largas y ovoideas, con bordes mal definidos y núcleo excéntrico en forma de grano de café. Presentan marcadores inmunohistoquímicos característicos (S100, CD1a, CD 45, HLA-DR, T200).2 La infiltración puede limitarse a un órgano o ser diseminada (afectación hepática, pulmonar, sistema hematopoyético), lo que condiciona el pronóstico.3 En general, los niños con afectación cutánea localizada evolucionan bien, pero un pequeño porcentaje puede evolucionar a afectación multisistémica, como en nuestro caso, lo que obliga a un seguimiento estrecho del paciente.4
1. Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127(22):2672-81. [ Links ]
2. Haupt R, Minkov M, Astigarraga I, et al. Langerhans Cell Histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013;60(2):175-84. [ Links ]
3. Singh A, Mandal A, Singh L, et al. Delayed Treatment Response in a Neonate with Multisystem Langerhans Cell Histiocytosis Case report and review of literature. Sultan Qaboos Univ Med J 2017;17(2):e225-8. [ Links ]
4. Donadieu J, Heritier S. Histiocytose langerhansienne de Tenfant. Presse Med 2017;46(1):85-95. [ Links ]
Presentación del nuevo caso clínico
En el próximo número se publicará el diagnóstico, manejo y tratamiento de este caso.
CASO CLÍNICO
Paciente de 11 años y 5 meses, con un cuadro de 30 días de evolución con epistaxis inicial, luego astenia, adinamia y disnea de esfuerzo. Sin otros antecedentes de relevancia previos.
En el examen físico, presentaba tensión arterial con valores superiores al percentil 95. Sudoroso. Palpación dolorosa en el hipocondrio derecho. Fondo de ojo normal. Al colocar el monitoreo multiparamétrico, se constata, en el electrocardiograma, el doble de la frecuencia cardíaca que la del oxímetro de pulso (Figura 1) y la del examen físico.
Radiografía de tórax con cardiomegalia. Cardiología informa electrocardiograma con frecuencia cardíaca (FC) de 110 latidos por minuto (lpm), PR 0,10", eje QRS +70, QRS alternante, aumento de voltaje de R en precordiales izquierdas, progresión RS izquierda y, en el ecocardiograma, miocardiopatía dilatada con disfunción leve del ventrículo izquierdo y fracción de acortamiento de 21%.
Análisis de laboratorio con resultados dentro de los parámetros normales. En la ecografía renal, se observa un tumor en ambas glándulas suprarrenales, que, junto con la tomografía computada de abdomen y la clínica, lleva al diagnóstico de feocromocitoma bilateral, confirmado en la cirugía, con evolución favorable y cese de la sintomatología.
¿Cuál es su diagnóstico?
• Pulso alternante.
• Pulso paradójico.
• Pulso en martillo de agua.
• Pulso alternante por bigeminismo extrasistólico.
• Disociación electromecánica 2 a 1.
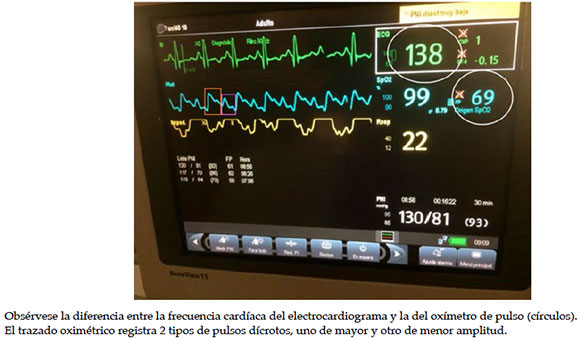
Figura 1. Foto del monitor del paciente
Para poder votar ingrese a: http://www.sap.org.ar/archivos

