










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.4 Buenos Aires Aug. 2018
http://dx.doi.org/10.5546/aap.2018.e603
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2018.e603
Monosomía 9p24 secundaria a translocación (2;9) en dos pacientes no relacionadas
Monosomy 9p24 in two non-related patients as result of a translocation (2;9)
Dra. Nayla Y. León-Carlosa, Dra. Constanza García-Delgadoa, Biol. Ariadna B. Morales-Jiméneza, Dr. Carlos Serrano-Bellob, M. en C. Alicia Cervantesc y Dra. Verónica F. Morán Barrosoa
a. Departamento de Genética, Hospital Infantil de México Federico Gómez.
b. Departamento de Patología, Hospital Infantil de México Federico Gómez.
c. Servicio de Genética, Hospital General de México Dr. Eduardo Liceaga/Facultad de Medicina, Universidad Nacional Autónoma de México, Ciudad de México, México.
Correspondencia: Dra. Verónica F. Morán Barroso: vfmoran@himfg.edu.mx
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 11-9-2017
Aceptado: 21-2-2018
RESUMEN
En pacientes con malformaciones congénitas y retraso del desarrollo psicomotor, deben descartarse cromosomopatías. Las más frecuentes son las translocaciones recíprocas balanceadas, presentes en 1:500 recién nacidos vivos. Por lo general, los portadores tienen fenotipo normal, aunque, ocasionalmente, presentan infertilidad, abortos o hijos con malformaciones. La translocación balanceada entre los cromosomas 2 y 9 puede originar descendencia con monosomías y trisomías de estos cromosomas. La monosomía del brazo corto del cromosoma 9 puede presentarse con trigonocefalia, dismorfias faciales, anomalías genitales y retraso del desarrollo psicomotor. En este trabajo, se revisaron las alteraciones de los cromosomas 2 y/o 9 en los cariotipos realizados en nuestra Institución en 2005-2014. Se presentan dos pacientes con monosomía 9p asociada a translocación (2;9). Las pacientes comparten datos de monosomía 9p24-pter; la correlación genotipo-fenotipo es compleja por el tamaño de los segmentos involucrados. Se resalta la importancia del diagnóstico cromosómico para el asesoramiento genético.
Palabras clave: Monosomía 9p; Translocación (2;9); Trisomía 2q; Anomalías congénitas; Bandeo cromosómico.
ABSTRACT
In patients with malformations and delayed psychomotor development it is important to discard chromosomopathies. Balanced reciprocal translocations are the most frequent chromosomopathies present in 1:500 live newborns. In general, carriers have normal phenotype, but they may have infertility, abortions or children with congenital malformations. The reciprocal translocation between chromosomes 2 and 9 can lead to offspring with monosomies and trisomies of these chromosomes. Short arm monosomy of chromosome 9 may present delayed psychomotor development, trigonocephaly, facial dysmorphia and genital abnormalities. We reviewed GTG karyotype records from our Institution to identify cases with chromosomes 2 and/or 9 alterations from 2005 to 2014. We describe two cases with monosomy 9p secondary to a translocation between chromosomes 2 and 9. The patients share features of monosomy 9p24-pter, however the genotype-phenotype correlation is complex due to the extension of the involved segments. We emphasize the importance of chromosomal diagnosis to offer genetic assessment.
Key words: Monosomy 9p; Translocation (2;9); Trisomy 2q; Congenital abnormalities; Chromosome banding.
INTRODUCCIÓN
En la consulta pediátrica, se evalúan pacientes con malformaciones congénitas y retraso en el desarrollo psicomotor (RDPM), alteraciones que pueden deberse a cromosomopatías.1 Las translocaciones recíprocas balanceadas, sin pérdida ni ganancia de material genético, son las aberraciones cromosómicas más frecuentes, presentes en 1:500 recién nacidos vivos.2 Por lo general, los portadores balanceados tienen fenotipo normal; sin embargo, pueden presentar infertilidad, abortos o descendencia con malformaciones congénitas por la formación de gametos desbalanceados. En ocasiones, los portadores tienen un fenotipo anormal, debido a un defecto monogénico por disrupción de un gen.3 La monosomía del brazo corto del cromosoma 9 (9p) es poco frecuente; se caracteriza por RDPM, trigonocefalia, dismorfias faciales y anomalías genitales.4,5 En el diagnóstico de las cromosomopatías, se utilizan diversos estudios, como el cariotipo con bandas G por tripsina y tinción con giemsa (GTG) o la hibridación in situ con fluorescencia (fluorescent in situ hy'ridization; FISH, por sus siglas en inglés), que detecta aberraciones en regiones específicas.
En este trabajo, se revisaron los resultados de cariotipos con bandas GTG de enero de 2005 a diciembre de 2014 para conocer la frecuencia de aberraciones estructurales de los cromosomas 2 y/o 9 en la población del Hospital Infantil de México Federico Gómez (HIMFG). Se reportaron 5762 cariotipos, con 22 resultados que involucraban los cromosomas 2 y/o 9 (0,38%). De ellos, tres tuvieron alteraciones del cromosoma 2 (una duplicación parcial del brazo largo -2q31-q33- y dos derivados de translocaciones); 16 mostraron alteración del cromosoma 9 y correspondieron a 8 derivados de una translocación, 5 deleciones, 3 de ellas de 9p; un anillo, una duplicación de brazo largo (9q) y uno con material adicional. Se identificó un caso que implicaba el cromosoma 2 y una inversión pericéntrica del cromosoma 9, y se expusieron dos casos con monosomía 9p secundaria a una translocación entre los cromosomas 2 y 9; se realizó un estudio clínico interdisciplinario y citogenético molecular.
DESCRIPCIÓN DE CASOS CLÍNICOS
Caso 1
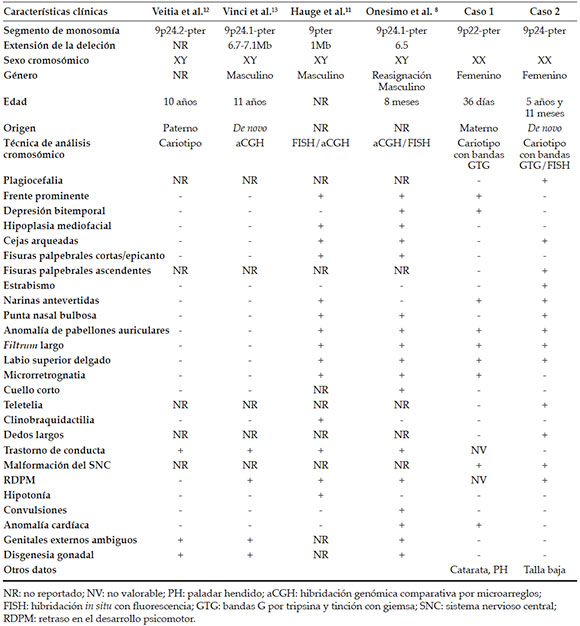
Recién nacida de padres jóvenes, sanos, no consanguíneos (Figura 1.A). Embarazo con esquema de maduración pulmonar con corticoesteroides por amenaza de parto pretérmino. Obtenida por cesárea a las 38 semanas de gestación con peso de 2550 g (percentil 3), talla de 45 cm (percentil 3) y puntaje de Apgar 6/8. Fue referida a los 6 días de vida por una cardiopatía congénita. Al momento de la exploración física, presentó braquicefalia, región frontal prominente, microftalmia y enoftalmos bilaterales, puente nasal deprimido, narinas antevertidas y cortas, paladar hendido central, pabellones auriculares dismórficos con hélix hipoplásico, soplo holosistólico y genitales externos femeninos (Figuras 1.B y 1.C). Se diagnosticó catarata congénita bilateral y coartación de aorta. Falleció a los 36 días de edad por complicaciones cardíacas.
El estudio post mórtem demostró agenesia del cuerpo calloso, comunicación interventricular y doble arteria renal bilateral (Tabla 1). El cariotipo mostró material adicional de origen desconocido en 9p, que fue 46,XX,add(9)(p22) (Figura 1.D). El cariotipo del padre fue normal y el de la madre fue 46,XX,t(2;9)(q31;p22), por lo que era portadora de una translocación balanceada entre los cromosomas 2 y 9 (Figura 1.E). El cariotipo final de la paciente fue 46,XX,der(9)t(2;9)(q31;p22) mat. El abuelo y el bisabuelo maternos resultaron portadores de la translocación y, en ellos, se realizó un análisis con FISH (ToTelVysion, Vysis, Abbott Park, ILL, USA) (Figura 1.F).

Figura 1. Caso 1.
Tabla 1. Características clínicas de la monosomía 9p24.3 reportadas y comparación con nuestras pacientes
Caso 2
Niña de 5 años y 11 meses de edad, tercera gestación de padres jóvenes, sanos, no consanguíneos; hermana de 11 años de edad con parálisis cerebral infantil. Embarazo con amenaza de aborto a los tres meses, infección de vías urinarias en el quinto mes y circular de cordón al cuello diagnosticada por ecografía obstétrica. Obtenida por cesárea a las 35 semanas de gestación con un peso de 2780 g (percentil 10), talla de 46 cm (percentil 10) y puntaje de Apgar 8/9. Referida por retraso de lenguaje afásico y moderado del desarrollo, atrofia cortical diagnosticada por resonancia magnética de encéfalo, talla baja y endotropia bilateral. Al momento de la exploración física, se encontró peso de 18,5 kg (percentil 10), talla de 107 cm (percentil 3), edad aparente menor que la cronológica, cráneo tendiente a la plagiocefalia, cejas pobladas y arqueadas, fisuras palpebrales ascendentes, punta nasal bulbosa, narinas antevertidas, hoyuelos retroauriculares izquierdos, filtrum largo, labios delgados, teletelia y genitales externos femeninos (Figuras 2.A y 2.B) (Tabla 1). El cariotipo reportó una translocación aparentemente balanceada entre los brazos cortos de los cromosomas 2 y 9: 46,XX,t(2;9) (p10;p24) (Figura 2.C). El cariotipo de ambos padres fue normal, por lo que la alteración se consideró de novo. El análisis por FISH con sondas subteloméricas de los cromosomas 2 y 9 en metafases de la propósita demostró la ausencia de la región subtelomérica 9p con cariotipo 46,XX,t(2;9)(p10;p24)del(9)(p24) (Figura 2.D), que correspondió a un rearreglo no balanceado: la región q31-qter del brazo largo del cromosoma 2 se translocó a la región distal del brazo corto del cromosoma 9 con deleción de la región p24-pter de este último.

Figura 2. Caso 2.
DISCUSIÓN
La frecuencia de alteraciones de los cromosomas 2 y 9 en el período analizado fue baja, con más alteraciones del 9 en una relación 4:1. La monosomía 9p presenta RDPM, trigonocefalia, puente nasal deprimido, filtrum largo, cuello corto y alado, y anomalías genitales. Las alteraciones genitales y/o de gónadas se han observado en individuos masculinos con la deleción de 9p24.3-pter, que incluye los genes DMRT.6 Establecer la correlación genotipo-fenotipo para la monosomía 9p es compleja por la variación en los puntos de ruptura y la participación de regiones trisómicas derivadas de otros cromosomas.7,8 La paciente 1 presentó malformaciones congénitas por ganancia de material genético en 2q31-2qter y pérdida en 9p22-9pter, alteraciones secundarias a la formación de un ovocito desbalanceado por segregación adyacente 1 de la translocación materna. La trisomía 2q presenta anormalidades craneofaciales (braquicefalia, región frontal prominente, hipertelorismo, puente nasal deprimido, narinas antevertidas, filtrum largo, pabellones auriculares de implantación baja, comisuras labiales descendentes, micrognatia) y alteraciones sistémicas, como RDPM, hipotonía, malformaciones cardíacas, urinarias y del tracto gastrointestinal.9 El fenotipo de la paciente correspondió a la trisomía 2q, con braquicefalia, región frontal prominente, comisuras labiales descendentes y alteraciones cardíacas y urinarias, que resultó letal por la extensión del material trisómico del cromosoma 2. La paciente también presentó fenotipo compatible con la monosomía 9p; sin embargo, no tuvo trigonocefalia, hipertelorismo, epicanto, cuello corto y alado, hipotonía ni alteraciones genitales.
En el segundo caso, la paciente tuvo una monosomía 9p24 pura. Las características en la paciente y en los casos ya reportados7,10-12 fueron RDPM, cejas arqueadas, narinas antevertidas, punta nasal bulbosa, anomalía de pabellones auriculares, filtrum largo y labio superior delgado. La paciente también presentó talla baja, plagiocefalia, fisuras palpebrales ascendentes, estrabismo, teletelia, dedos largos y malformación del sistema nervioso central.
En los dos casos aquí descritos, hubo pérdida de 9p24, región relacionada con alteraciones del desarrollo sexual por genes sensibles a dosis, como DMRT, necesarios para la formación de testículos en individuos XY y para la función ovárica en individuos XX.12 DMTR tiene expresión gonadal sexual específica; el desarrollo testicular alterado y la feminización XY ocurren cuando este gen se encuentra en estado hemicigoto.13 En las pacientes expuestas, no hubo alteraciones genitales y/o gonadales, lo que podría deberse a que estas son más frecuentes en individuos masculinos.6,14
No se ha encontrado alguna característica clínica presente en todos los casos de monosomía 9p24.7,10-12 Las pacientes expuestas compartían la malformación del sistema nervioso central, narinas antevertidas, anomalías en pabellones auriculares y filtrum largo. Su comparación es complicada, ya que, en el primer caso, la región perdida de 9p era más extensa y su fenotipo se debía en forma predominante a la trisomía 2q31-qter. Es interesante que, en esta revisión, se hayan identificado dos casos de monosomía 9p secundarias a una translocación con el cromosoma 2, pues son poco frecuentes.6,7,10-12,15
El asesoramiento genético del caso 1 con madre portadora balanceada de t(2;9) incluyó el riesgo teórico por embarazo de presentar abortos (33%), hijos con malformaciones por monosomías o trisomías relacionadas con la translocación (33%), portadores balanceados de la translocación sanos en fenotipo (16%) o sanos en genotipo y fenotipo (16%). El riesgo de recurrencia para el caso 2 fue similar al de la población general por ser de novo.
CONCLUSIONES
Las alteraciones estructurales de los cromosomas 2 y/o 9 en nuestra Institución fueron pocas, con mayor frecuencia en el cromosoma 9 y derivados de translocaciones recíprocas. El análisis de nuestros casos amplía la correlación genotipo-fenotipo de la monosomía 9p con nuevos datos clínicos, como talla baja, plagiocefalia, fisuras palpebrales ascendentes, estrabismo, teletelia, dedos largos y malformación del sistema nervioso central. Se resalta la importancia del diagnóstico cromosómico para el manejo y asesoramiento genético de estos casos.
1. Martin CL, Ledbetter D. Chromosomal Microarray T esting for Children with Unexplained Neurodevelopmental Disorders. JAMA 2017; 317(24):2545. [ Links ]
2. Kaiser-Rogers K, Rao KW. Structural chromosome rearrangements. In: Gersen S, Keagle M (eds.). The Principies of Clinical Cytogenetics. 3th ed. New York: Springer; 2013. P. 159-61. [ Links ]
3. Midro AT, Zollino M, Wiland E, et al. Meiotic and pedigree segregation analyses in carriers of t(4;8)(p16;p23.1) differing in localization of breakpoint positions at 4p subband 4p16.3 and 4p16.1. J Assist Reprod Genet 2016; 33(2):189-97. [ Links ]
4. Swinkels ME, Simons A, Smeets DF, et al. Clinical and cytogenetic characterization of 13 Dutch patients with deletion 9p syndrome: Delineation of the critical region for a consensus phenotype. Am J Med Genet A 2008; 146A(11):1430-8. [ Links ]
5. Durmaz CD, Yararbaş K, Kutlay NY, et al. Unusual Chromosomal Rearrangement Resulted in Interstitial Monosomy 9p: Case Report. Cytogenet Genome Res 2016; 148(1):19-24.
6. Barbaro M, Balsamo A, Anderlid BM, et al. Characterization of deletions at 9p affecting candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur JMed Genet 2009; 17(11):1439-47. [ Links ]
7. Onesimo R, Orteschi D, Scalzone M, et al. Chromosome 9p deletion syndrome and sex reversal: Novel findings and redefinition of the critically deleted regions. Am J Med Genet A 2012; 158A(9):2266-71. [ Links ]
8. Sirisena ND, Wijetunge UK, de Silva R, et al. Child with deletion 9p syndrome presenting with craniofacial dysmorphism, developmental delay, and multiple congenital malformations. Case Rep Genet 2013; 2013:785830. [ Links ]
9. Angle B, Hersh JH, Yen F, et al. Case of partial duplication 2q3 with characteristic phenotype: rare occurrence of an unbalanced offspring resulting from a parental pericentric inversion. Am J Med Genet 2000; 91(2):126-30. [ Links ]
10. Hauge X, Raca G, Cooper S, et al. Detailed characterization and clinical correlations in 10 patients with distal deletions of chromosome 9p. Genet Med 2008; 10(8):599-611. [ Links ]
11. Veitia RA, Nunes M, Quintana-Murci L, et al. Swyer syndrome and 46,XY partial gonadal dysgenesis associated with 9p deletions in the absence of monosomy-9p syndrome. Am J Hum Genet 1998; 63(3):901-5. [ Links ]
12. Vinci G, Chantot-Bastaraud S, El Houate B, et al. Association of deletion 9p, 46, XY gonadal dysgenesis and autistic spectrum disorder. Mol Hum Reprod 2007; 13(9):685-9. [ Links ]
13. Smith CA, McClive PJ, Western PS, et al. Conservation of a sex-determining gene. Nature 1999; 402(6762):601-2. [ Links ]
14. Abreu LS, Brassesco MS, Moreira ML, et al. Case report. Familial balanced translocation leading to an offspring with phenotypic manifestations of 9p syndrome. Genet Mol Res 2014; 13(2):4302-10. [ Links ]
15. Ledig S, Hiort O, Scherer G, et al. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: Evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod 2010; 25(10): 2637-46. [ Links ]

