










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.5 Buenos Aires Oct. 2018
http://dx.doi.org/10.5546/aap.2018.368
ACTUALIZACIÓN
http://dx.doi.org/10.5546/aap.2018.368
Síndrome de Beckwith-Wiedemann. Aspectos clínicos y etiopatogénicos de una entidad ejemplo de impronta genómica
Prof. Francisco Cammarata-Scalisia, Prof. Andrea Avendañoa, Dra. Frances Stockb, Dr. Michele Calleac, Dra. Angela Sparagod y Prof. Andrea Ricciod,e
a. Unidad de Genética Médica, Departamento de Puericultura y Pediatría, Universidad de Los Andes, Mérida, Venezuela.
b. Unidad de Oncología Pediátrica, Instituto Autónomo Hospital Universitario de Los Andes, Mérida, Venezuela.
c. Unit of Dentistry, Bambino Gesù Children's Hospital -IRCCS, Rome, Italy.
d. Department of Environmental, Biological and Pharmaceutical Sciences and Technologies, University of Campania "Luigi Vanvitelli", Caserta, Italy.
e. Institute of Genetics and Biophysics "Adriano Buzzati-Traverso", Consiglio Nazionale delle Ricerche (CNR), Naples, Italy.
Correspondencia: Prof. Francisco Cammarata-Scalisi: francocammarata19@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 18-10-2017
Aceptado: 8-3-2018
RESUMEN
El síndrome de Beckwith-Wiedemann es la entidad genética de sobrecrecimiento más común, con una incidencia aproximada de 1 en 10 000-13 700 nacimientos. Presenta un amplio espectro clínico, que incluye macrosomía pre- y posnatal, macroglosia, alteraciones en el pabellón auricular, defectos en la pared abdominal, visceromegalia e hipoglucemia por hiperinsulinemia. Es un síndrome de predisposición a cáncer en la infancia, causado por una variedad de alteraciones genéticas y/o epigenéticas que suelen afectar la regulación de los genes impresos en 11p15.5. Conocer las correlaciones (epi) genotipo/fenotipo ha impulsado recomendaciones para plantear las diferentes estrategias de atención, entre ellas, los protocolos de vigilancia de tumores basados en la clasificación molecular, con la finalidad de estandarizar la práctica clínica. El objetivo del presente artículo es mostrar el estado actual del síndrome de Beckwith-Wiedemann, un ejemplo de impronta genómica.
Palabras clave: Síndrome de Beckwith-Wiedemann; Neoplasias; Predisposición genética a la enfermedad; Impresión genómica; Correlaciones genotipo-fenotipo.
INTRODUCCIÓN
El síndrome de Beckwith-Wiedemann (SBW, OMIM 130650) es la entidad genética de sobrecrecimiento más común.1-9 Fue descrito por primera vez por Beckwith en 1963 y Wiedemann en 1964.5 Es panétnico, con una razón por sexo de 1:1,2 y una incidencia aproximada de 1 en 10 000-13 700 nacimientos.1,2,8
Esta cifra se puede subestimar en los casos de fenotipos leves y es posible que aumente debido a una correlación positiva al uso de técnicas de reproducción asistida.8
CLÍNICA Y SEGUIMIENTO
Presenta un variable y amplio espectro clínico, que incluye los antecedentes de polihidramnios7 y macrosomía prenatal.2-7 Posteriormente, se caracteriza por crecimiento posnatal excesivo,5 hipotonía,7 hemangiomas, nevus flammeus en la glabela,1,4,5,8-11 pliegue infraorbiatrio,11 hipoplasia mediofacial,8,11 macroglosia,1-11 paladar hendido,4,8,9,11 sialorrea, prognatismo,8,11 alteraciones en el pabellón auricular (pliegues en el lóbulo e indentaciones posteriores del hélix, Figura 1),,1,4,8,11 disnea,8 anomalías cardíacas4 (cardiomegalia e, infrecuentemente, el síndrome de QT largo),8 pezones supernumerarios,11 defectos en la pared abdominal (onfalocele, hernia umbilical1-9 y diástasis de los rectos),8,10 visceromegalia3,5,8,10,12 (hígado, páncreas, bazo o riñones),3,12,13 riñón con médula en esponja,12 malformaciones urorrenales,3,4,5 hemihiperplasia corporal,1-3,5,6,9,10,13 comúnmente evidente al momento del nacimiento,8 polidactilia4,11 e hipoglucemia por hiperinsulinemia,8,10 entre otras características fenotípicas,3,6,9 y el desarrollo psicomotor suele ser normal.11
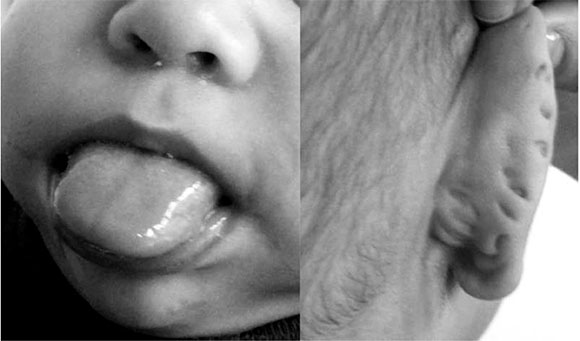
Figura 1. Macroglosia y alteraciones en el pabellón auricular (Mentaciones posteriores del hélix)
El diagnóstico se basa en signos clínicos, y la presencia de tres signos mayores o dos mayores y uno menor puede orientar el diagnóstico clínico (Tabla 1)1,4,10,11
Tabla 1. Criterios diagnósticos del síndrome de Beckwith-Wiedemann1,4,10,11

Ante la presencia de macrosomía, puede existir un mayor riesgo de traumatismos en los recién nacidos con cefalohematoma, lesión en el plexo braquial, síndrome de dificultad respiratoria y hasta producirse la muerte. Estas posibles complicaciones aumentan la probabilidad de optar por la realización de cesárea y la asociación con el parto prematuro ha sido encontrada.11
Por su parte, la macroglosia es la característica más común. Se puede encontrar hasta en 97% de los pacientes (Tabla 1). Constituye el rasgo más sensible para la identificación de la entidad11 y un factor de riesgo para la apnea obstructiva del sueño. El tratamiento quirúrgico de reducción ha sido exitoso en este trastorno, aunque es necesario comprender mejor cuáles individuos se pueden beneficiar de este procedimiento14 y su impacto en la respiración,15 el lenguaje y la deglución.14,15 Presenta, además, otras implicaciones estéticas caracterizadas por la apariencia de boca abierta, aumento en los espacios interdentales y prognatismo, que puede causar consecuencias negativas en relación con la imagen corporal y la alteración del bienestar psicológico.15 De forma infrecuente, se puede presentar hipoacusia de tipo conductiva, que debe manejarse para evitar trastornos en el aprendizaje.8
En el caso de la hipoglucemia, se debe descartar el hiperinsulinismo y la nesidioblastosis, por lo que se considera la evaluación por Endocrinología Infantil y la implementación de medicación temprana,16 y, con ello, se busca reducir las convulsiones que pueden conducir al retraso en el desarrollo.17
Es un síndrome de predisposición a cáncer en la infancia, en comparación con la población general,1-3,5,7,910 con riesgo de malignidad estimado de 4% a 21% (~7,5%).2,4,8 Es mayor al nacer y se aproxima a la línea de base de la población general antes de la pubertad.1 El espectro de tumores comprende, principalmente, histiotipos embrionarios, como el tumor de Wilms1,2,4,6,8,13 y el hepatoblastoma, entre los más frecuentes,1,4 además de neuroblastoma,4,8 carcinoma adrenocortical,4,18 feocromocitomas19 y rabdomiosarcomas.68 Sin embargo, el riesgo de aparición de tumores difiere considerablemente entre la expresión anormal de un grupo de genes con impronta localizados en 11p15.5.34
El reconocimiento precoz durante el período prenatal o neonatal es esencial, ya que facilita las intervenciones médico-quirúrgicas que puedan cubrir las complicaciones presentadas, mientras que se inicia el monitoreo a largo plazo para la detección de las neoplasias anteriormente mencionadas, por lo que se permite educar a los padres sobre los diversos tratamientos.
DIAGNÓSTICO PRENATAL
La evaluación prenatal se recomienda en circunstancias en las que existe una historia familiar positiva o se diagnostican algunas características clínicas de la entidad. Se confirma por la presencia de dos características mayores, (macroglosia, macrosomía, defectos en la pared abdominal, como el onfalocele, placentomegalia) o una característica mayor y dos características menores, (polihidramnios, nefromegalia, displasia o citomegalia suprarrenal). Estos pueden ser explorados por ecografía a las 18-20 semanas de gestación y confirmar a las 25-32 semanas de gestación.8 Es por esto por lo que se debe considerar la realización de una ecografía morfogenética que evalúe con detalle la región craneofacial y el abdomen, en la que se incluya la medición de órganos sólidos. Además, la elevación de niveles de alfa-fetoproteína sérica se asocia con la presencia de onfalocele.16
La realización de la biopsia de vellosidades coriónicas en el primer trimestre o de amniocentesis en el segundo trimestre puede ser empleada para determinar las diferentes alteraciones moleculares, que se expondrán posteriormente. Durante la gestación, se puede desarrollar mayor riesgo de hipertensión y proteinuria (sugestivo de preeclampsia) y diabetes mellitus gestacional.8
FACTORES DE RIESGO
La frecuencia en gemelos monocigóticos entre los individuos con SBW es más alta que en la población general (2,5% versus 0,3-0,4%)89 y más frecuente en el tipo monocorial diamniótico.9 Es predominante en el sexo femenino8 y en gemelos, en los que uno muestra el fenotipo de la entidad y el otro muestra un fenotipo normal o parcial.9
Desde el primer caso de SBW concebido a través de técnicas de reproducción asistida en 1995,10 se ha asociado a un mayor riesgo ante el empleo de estas herramientas y la presencia de defectos de impronta genómica.7 La incidencia del SBW en la población concebida por esta técnica es aproximadamente 1 en 4000 nacimientos.1 Durante el desarrollo embrionario temprano, el mecanismo epigenético puede alterar los patrones de metilación, después de la implantación del blastocisto hasta el final del desarrollo del embrión, cuando se añaden marcas de metilación del ácido desoxirribonucleico (ADN) en aquellas regiones susceptibles de regulación. Indistintamente, los pacientes con problemas de fertilidad tratados o no tienen una mayor frecuencia de trastornos de impresión y es por ello por lo que se ha sugerido a estas técnicas como un factor de riesgo.7
DIAGNÓSTICOS DIFERENCIALES
Se encuentran otras entidades de sobrecrecimiento, que incluyen los síndromes de Sotos, Simpson-Golabi-Behmel, Costello8,20,21 y Perlman,20,21 y se puede dificultar la distinción en el período prenatal. Se deben descartar otras entidades endócrinas, como el hipotiroidismo congénito, alteraciones metabólicas con dismorfismo facial, como las mucopolisacaridosis (síndromes de Hurler, Hunter y Maroteaux-Lamy), además de las gangliosidosis y enfermedad de Pompe.1
ETIOPATOGENIA
El SBW es causado por una variedad de alteraciones genéticas y/o epigenéticas que suelen afectar la regulación de los genes impresos en 11p15.5,26 que pueden producir heterogeneidad en el espectro clínico. Es, por lo tanto, un paradigma de alteraciones congénitas asociadas con la impronta genómica,1,5 un proceso que consiste en la expresión génica específica de origen parental. Hasta 90% de los casos es causado por la alteración en la expresión de genes implicados en la progresión del ciclo celular y control del crecimiento somático, regulada por dos centros de impronta independientes (CI1 y CI2),1,6,7,22 que abarcan aproximadamente 1 Mb.7
En todo el genoma, hay aproximadamente 120 genes impresos que están asociados con 44 centros de impronta. Además del SBW, otros trastornos han sido bien caracterizados, como el síndrome de Prader-Willi, Angelman, Temple, Kagami-Ogata y Silver-Russell, junto con la diabetes mellitus neonatal transitoria y el pseudohipoparatiroidismo.7
La impronta se refiere a la expresión preferencial o exclusiva del alelo paterno o materno de un gen impreso. La expresión génica de impronta está regulada por mecanismos epigenéticos, y la más común es la metilación del ADN de los centros de impronta ricos en islas CpG, que conforman un porcentaje importante en los promotores de genes.2 El CI1 y CI2 son caracterizados por metilaciones diferenciales de sus alelos maternos y paternos,1 y en condiciones normales, el CI1 del alelo paterno y el CI2 del alelo materno están metilados.5
La principal causa se debe a la pérdida de metilación en CI2, en 50-60% de los casos (Tabla 2), el cual es centromérico en relación con CI1.1,2,4,5,9,10,22 Produce la reducción de la expresión del gen inhibidor de cinasa dependiente de ciclina 1C (CDKN1C),1,22 que funciona como gen supresor de tumor y un regulador negativo del crecimiento fetal,2 normalmente expresado por el cromosoma materno.1,22 Los individuos portadores de este tipo de alteración genética son más propensos a presentar macroglosia,14 hepatoblastoma, neuroblastoma y tumores suprarrenales.12
Tabla 2. Factores etiopatogénicos en el síndrome de Beckwith-Wiedemann, frecuencia y hallazgos asociados1,2,4,7,9,13,17,31,32
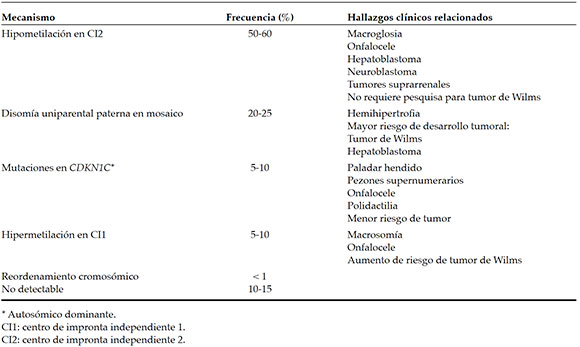
Además, las mutaciones puntuales maternas en CDKN1C representan 5-10% de los casos1,4,5,6,9,10 y son responsables de 5% de los casos esporádicos1,4 y la mitad de los casos con historia familiar positiva,1,6,7 con preferencia materna y patrón de herencia autosómico dominante.21 Es por ello por lo que la mayoría de los casos del SBW son esporádicos.10,21 La polidactilia, los pezones supernumerarios y el paladar hendido se observan con mayor frecuencia en este tipo de alteración génica.11
El onfalocele ocurre con mayor frecuencia en pacientes con hipometilación de CI22,6 o mutaciones puntuales en el gen CDKN1C.2,6,13 No obstante, el riesgo de tumor es significativamente menor ante estas dos circunstancias,2,6 y múltiples investigaciones han concluido que la pérdida de metilación en CI2 no confiere un mayor riesgo y, por lo tanto, no requieren pesquisa para el tumor de Wilms.2
Además, 5-10% de los casos son causados por hipermetilación en CI1,1,4,5,10,17 que da como resultado la regulación de la expresión bialélica del gen del factor de crecimiento similar a la insulina tipo 2 IGF2, normalmente expresado en el alelo paterno, y el ácido ribonucleico (ARN) no codificante del gen oncosupresor H19, normalmente expresado por el alelo materno.1,2,4,22 La presencia de macrosomía,4 onfalocele13 y un mayor riesgo de tumor de Wilms se asocia más comúnmente a este tipo de alteración epigenética.4,13
La alteración en la metilación de CI1 y CI2 se explica por una disomía uniparental paterna en mosaico, que ocurre en 20-25%1,2,5,6,9,10 y se encuentra asociada con características fenotípicas adicionales,1 como la hemihipertrofia,4,13 y un riesgo más alto de desarrollo tumoral, especialmente para tumor de Wilms2,6,12 y el hepatoblastoma.13,23
En general, menos de 1% de los casos son causados por reordenamientos cromosómicos, tales como duplicaciones, translocaciones, inversiones, deleciones, que engloban los genes en el grupo de CI,1,5,22 y alrededor de 10-15% de los individuos clínicamente diagnosticados no tienen defecto molecular detectable, a pesar de presentar un fenotipo evidente.1,2,5,6
PROTOCOLOS DE VIGILANCIA DE TUMORES
Aunque todos estos mecanismos juegan aún un papel desconocido en la patogénesis del SBW,6 conocer las correlaciones (epi)genotipo/fenotipo ha impulsado recomendaciones para plantear las diferentes estrategias de atención, entre ellas, los protocolos de vigilancia de tumores basados en la clasificación molecular, con el objetivo de estandarizar la práctica clínica.511
Ante los hallazgos genéticos anteriormente expuestos, se debe realizar una ecografía abdominal (cada 3-4 meses en la infancia para descartar el tumor de Wilms6,23 y el neuroblastoma). Otros estudios que pueden formar parte de esta pesquisa son la radiografía de tórax anual y la tomografía computarizada abdominal si existe nefromegalia o alguna imagen sospechosa.16 Además, el cribado de niveles de alfa-fetoproteína sérica (cada 2-3 meses durante los primeros cuatro años) para la pesquisa del hepatoblastoma.6,16,23 Igualmente, se deben determinar los valores de gonadotropina coriónica y catecolaminas para la detección de tumores de células germinales y neuroblastoma, respectivamente. Los estudios de orina completa anual también deben formar parte de la evaluación. La detección temprana puede garantizar el adecuado manejo de estas neoplasias malignas de alto riesgo.16 Se debe promover este cribado, en especial, en los casos de disomía uniparental paterna.23
ASESORAMIENTO GENÉTICO
Como ya se hizo referencia, la mayoría de los pacientes con SBW son esporádicos, por lo que el caso estudiado sería, por lo general, el único afectado en el grupo familiar. Excepto cuando se determina mutación puntual en el gen CDKN1C, que exhibe un patrón de herencia autosómica dominante, por lo que el riesgo de recurrencia es de 50% en los que presentan dicha alteración genética. El pronóstico varía de acuerdo con la forma de presentación de la entidad, la cual puede ser diferente incluso en los casos familiares.
Por lo tanto, el seguimiento y control de estos pacientes debe iniciarse de forma precoz e individual con el objeto de minimizar las complicaciones que pueden presentarse y brindar el tratamiento disponible.
El objeto del presente artículo es mostrar el estado actual del SBW, poder realizar el diagnóstico a través de una aproximación clínica y descartar los diagnósticos diferenciales. Las causas etiopatogénicas son variadas y es necesario comprenderlas para orientar los protocolos de vigilancia médicos interdisciplinarios, que deben ser individualizados e incluir un oportuno asesoramiento genético familiar.
1. Mussa A, Russo S, De Crescenzo A, et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet 2016; 24(2):183-90. [ Links ]
2. Brzezinski J, Shuman C, Choufani S, et al. Wilms tumour in Beckwith-Wiedemann syndrome and loss of methylation at imprinting centre 2: revisiting tumour surveillance guidelines. Eur J Hum Genet 2017; 25(9):1031-9. [ Links ]
3. Giabicani É, Brioude F, Le Bouc Y, et al. Imprinted disorders and growth. Ann Endocrinol (Paris) 2017; 78(2):112-3. [ Links ]
4. Lin HY, Chuang CK, Tu RY, et al. Epigenotype, genotype, and phenotype analysis of patients in Taiwan with Beckwith-Wiedemann syndrome. Mol Genet Metab 2016; 119(1-2):8-13. [ Links ]
5. Luk HM. Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population. J Pediatr Endocrinol Metab 2017; 30(1):89-95. [ Links ]
6. Maas SM, Vansenne F, Kadouch DJ, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am JMed Genet A 2016; 170(9):2248-60. [ Links ]
7. Tenorio J, Romanelli V, Martin-Trujillo A, et al. Clinical and molecular analyses of Beckwith-Wiedemann syndrome: Comparison between spontaneous conception and assisted reproduction techniques. Am J Med Genet A 2016; 170(10):2740-9. [ Links ]
8. Zammit M, Caruana E, Cassar D, et al. Beckwith-Wiedemann Syndrome Review: A Guide for the Neonatal Nurse. Neonatal Netw 2017; 36(3):129-33. [ Links ]
9. Inoue T, Nakamura A, Matsubara K, et al. Continuous hypomethylation of the KCNQ1OT1:TSS-DMR in monochorionic twins discordant for Beckwith-Wiedemann syndrome. Am J Med Genet A 2017; 173(10):2847-50. [ Links ]
10. Mussa A, Molinatto C, Cerrato F, et al. Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics 2017; 140(1):e20164311. [ Links ]
11. Mussa A, Di Candia S, Russo S, et al. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur J Med Genet 2016; 59(1):52-64. [ Links ]
12. Cheungpasitporn W, Erickson SB. Beckwith-Wiedemann syndrome and recurrent bilateral renal calculi. Urol Ann 2017; 9(1):113-4. [ Links ]
13. Eggermann K, Bliek J, Brioude F, et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. Eur J Hum Genet 2016; 24(10):1377-87. [ Links ]
14. Cielo CM, Duffy KA, Vyas A, et al. Obstructive sleep apnoea and the role of tongue reduction surgery in children with Beckwith-Wiedemann syndrome. Paediatr Respir Rev 2018; 25:58-63. [ Links ]
15. Maas SM, Kadouch DJ, Masselink AC, et al. Taste and speech following surgical tongue reduction in children with Beckwith-Wiedemann síndrome. J Craniomaxillofac Surg 2016; 44(6):659-63. [ Links ]
16. Lapunzina Badía P, del Campo Casanelles M, Delicado Navarro A, et al. Guía clínica para el seguimiento de pacientes con síndrome de Beckwith-Wiedemann. An Pediatr (Bare) 2006; 64(3):252-9. [ Links ]
17. Spivey PS, Bradshaw WT. Recognition and management of the infant with Beckwith-Wiedemann Syndrome. Adv Neonatal Care 2009; 9(6):279-84. [ Links ]
18. MacFarland SP, Mostoufi-Moab S, Zelley K, et al. Management of adrenal masses in patients with Beckwith-Wiedemann syndrome. Pediatr Blood Cancer 2017; 64(8):26432. [ Links ]
19. Caza T, Manwaring J, Riddell J. Recurrent, bilateral, and metastatic pheochromocytoma in a young patient with Beckwith-Wiedemann syndrome: A genetic link? Can Urol Assoc J 2017; 11(5):e240-3. [ Links ]
20. Callea M, Yavuz I, Clarich G, et al. A case of Beckwith-Wiedemann syndrome with peculiar dental findings. Eur J Paediatr Dent 2016; 17(4):315-7. [ Links ]
21. Ferianec V, Bartova M. Beckwith-Wiedemann syndrome with overlapping Perlman syndrome manifestation. J Matern Fetal Neonatal Med 2014; 27(15):1607-9. [ Links ]
22. Mussa A, Russo S, de Crescenzo A, et al. Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin Genet 2016; 90(1):21-7. [ Links ]
23. Mussa A, Ferrero GB. Serum alpha-fetoprotein screening for hepatoblastoma in Beckwith-Wiedemann syndrome. Am J Med Genet A 2017; 173(3):585-7. [ Links ]

