










Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.5 Buenos Aires oct. 2018
http://dx.doi.org/10.5546/aap.2018.368
REVIEW
http://dx.doi.org/10.5546/aap.2018.eng.368
Beckwith-Wiedemann syndrome. Clinical and etiopathogenic aspects of a model genomic imprinting entity
Prof. Francisco Cammarata-Scalisia, Prof.Andrea Avendañoa, Frances Stock, M.D.b, Michele Callea, M.D.c, Angela Sparago, PhD and Prof. Andrea Ricciod,e
a. Unit of Medical Genetics, Department of Newborn Infant Care and Pediatrics, Universidad de Los Andes, Mérida, Venezuela.
b. Unit of Pediatric Oncology, InstitutoAutónomo Hospital Universitario de Los Andes, Mérida, Venezuela.
c. Unit of Dentistry, Bambino Gesù Children's Hospital -IRCCS, Rome, Italy.
d. Department of Environmental, Biological and Pharmaceutical Sciences and Technologies, University of Campania "Luigi Vanvitelli," Caserta, Italy.
e. Institute of Genetics and Biophysics "Adriano Buzzati-Traverso," Consiglio Nazionale delle Ricerche (CNR), Naples, Italy.
E-mail address: Prof. Francisco Cammarata-Scalisi: francocammarata19@ gmail.com
Funding: None.
Conflict of interest: None.
Received: 10-18-2017
Accepted: 3-8-2018
ABSTRACT
The Beckwith-Wiedemann syndrome is the most common genetic entity in overgrowth, with an approximate incidence of 1 in 10 00013 700births. Its broad clinical spectrum includes pre- and postnatal macrosomia, macroglossia, pinna abnormalities, abdominal wall defects, visceromegaly, and hyperinsulinemic hypoglycemia. This syndrome predisposes to childhood cancer and is caused by diverse genetic and/or epigenetic disorders that usually affect the regulation of genes imprinted on chromosome 11p15.5. The knowledge of (epi) genotype-phenotype correlations has prompted recommendations to propose different health care strategies, including tumor surveillance protocols based on molecular classification, aimed at standardizing clinical practice. The objective of this article is to describe the current status of the Beckwith-Wiedemann syndrome, a model of genomic imprinting.
Key words: Beckwith-Wiedemann syndrome; Neoplasias; Genetic predisposition to disease; Genomic imprinting; Genotype-phenotype correlations.
INTRODUCTION
The Beckwith-Wiedemann syndrome (BWS, OMIM #130650) is the most common genetic entity in overgrowth.1-9 It was first described by Beckwith in 1963 and Wiedemann in 1964.5 It is a panethnic syndrome with a 1:1,2 sex ratio and an approximate incidence of 1 in 10 00013 700 births.1,2,8 Such incidence may be underestimated in mild phenotypes and most likely increases due to a positive correlation with assisted reproductive techniques.8
CLINICAL FEATURES AND FOLLOW-UP
The clinical spectrum is wide and varied and includes a history of polihydramnios7 and prenatal macrosomia.2-7 At a later stage, it is characterized by postnatal overgrowth,5 hypotonia,7 hemangiomas, nevus flammeus of the forehead,1,4,5,8-11 infraorbital fold,11 midfacial hypoplasia,8,11 macroglossia,1-11 cleft palate,4,8,9,11 ptyalism, prognathism,8,11 pinna abnormalities (ear lobe creases and posterior helical ear pits, Figure 1),1,4,8,11 dyspnea,8 heart anomalies4 (cardiomegaly and, rarely, long QT syndrome),8 supernumerary nipples,11 abdominal wall defects (omphalocele, umbilical hernia,1-9 and diastasis recti),8,10 visceromegaly3,5,8,10,12 (liver, pancreas, spleen or kidneys),3,12,13 medullary sponge kidney,12 malformations of the kidney and ureter,3,4,5 whole-body hemihypertrophy,1-3,5,6,91013 which is commonly evident at birth,8 polydactyly,4,11 and hyperinsulinemic hypoglycemia,8,10 among other phenotypic features,3,6,9 although psychomotor development is usually normal.11
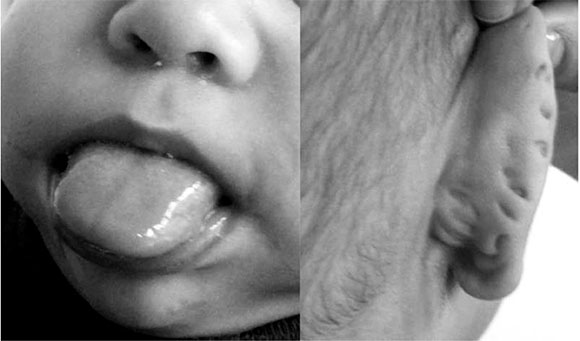
Figure 1. Macroglossia and pinna abnormalities (posterior helical ear pits)
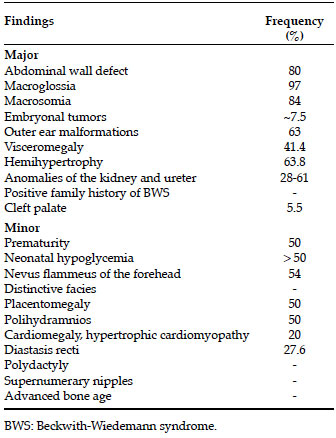
The diagnosis is based on clinical signs, and the presence of three major signs or two major and one minor sign may guide clinical diagnosis (Table 1).1,4,10,11
Table 1. Diagnostic criteria of Beckwith-Wiedemann syndrome1,4,10,11
In the presence of macrosomia, there may be a greater risk for trauma among newborn infants, such as cephalohematoma, brachial plexus injury, respiratory distress syndrome, and even death. Such potential complications increase the probability of choosing to perform a cesarean section, and an association has been established with preterm birth.11
On its side, macroglossia is the most common feature. It may be observed in up to 97% of patients (Table 1). It is the most easily identifiable feature of BWS11 and a risk factor for obstructive sleep apnea. Surgical reduction management has been successful in macroglossia, although it is necessary to better understand which individuals may benefit from this procedure14 and its impact on breathing,15 language, and swallowing.14,15 It also involves other aesthetic aspects, such as open mouth posture, increased interdental space, and prognathism, which may have negative consequences in relation to body image and an altered psychological well-being.15 Infrequently, conductive hearing loss may occur, which should be managed to avoid learning disorders.8
In the case of hypoglycemia, hyperinsulinemia and nesidioblastosis should be ruled out, so an assessment by the Pediatric Endocrinology Service and an early drug treatment should be considered,16 in order to reduce seizures that may lead to developmental delay.17
BWS predisposes to childhood cancer, compared to the general population,1-3,5,7,9,10 with an estimated risk for malignancy of 4-21% (~7.5%).2,4,8 The risk is higher at birth and reaches the general population's baseline before puberty.1 Tumors mainly include embryonal histiotypes, such as Wilms tumor1,2,4,6,8,13 and hepatoblastoma, among the most common ones,1,4 in addition to neuroblastoma,4,8 adrenocortical carcinoma,4,18 pheochromocytomas,19 and rhabdomyosarcomas.6,8 However, the risk for tumors is significantly different from the abnormal expression of a group of genes imprinted on chromosome 11p15.5.3,4
The early recognition during the prenatal or neonatal period is critical because it facilitates medical-surgical interventions that may cover any observed complication, while starting long-term monitoring for the neoplasias mentioned above, which allows to educate parents on the different treatments available.
PRENATAL DIAGNOSIS
Prenatal assessment is recommended when there is a positive family history or certain BWS clinical features are diagnosed. It is confirmed by the presence of two major features (macroglossia, macrosomia, abdominal wall defects, such as omphalocele, placentomegaly) or one major and two minor features (polihydramnios, nephromegaly, adrenal dysplasia or cytomegaly). These may be explored by ultrasound around 18-20 weeks of gestation and then confirmed at 25-32 weeks of gestation.8 For this reason, it is necessary to consider performing a morphogenetic assessment of the fetus using ultrasound to establish the status of its abdominal and craniofacial regions, including the measurement of solid organs. In addition, high serum alpha-fetoprotein levels are associated with the presence of omphalocele.16
Chorionic villus sampling in the first trimester or an amniocentesis in the second trimester may help to determine the different molecular alterations that will become exposed subsequently. During gestation, there may be a higher risk for hypertension and proteinuria (suggestive of preeclampsia), and gestational diabetes mellitus.8
RISK FACTORS
The frequency of monozygotic twins among patients with BWS is higher than in the general population (2.5% compared to 0.3-0.4%)8,9 and it is more common in the monochorionic diamniotic type.9 It predominates among female individuals8 and twins, where one has the BWS phenotype and the other one has a normal or partial phenotype.9
Since the first BWS case conceived using assisted reproductive techniques in 1995,10 it has been associated with a higher risk when using these tools and the presence of genomic imprinting defects.7 The incidence of BWS in the population conceived using these techniques is approximately 1 in 4000 births.1 During early embryonic development, the epigenetic mechanism may alter methylation patterns, after the blastocyst implantation until the end of the embryo development, when deoxyribonucleic acid (DNA) methylation marks are added in regulation-susceptible regions. Without distinction, patients with fertility problems, either treated or not, have a higher frequency of imprinting disorders; for this reason, it has been suggested that these techniques are a risk factor.7
DIFFERENTIAL DIAGNOSES
There are other overgrowth entities, including the Sotos, Simpson-Golabi-Behmel, Costello,8,20,21 and Perlman syndromes,20,21 which may hinder the possibility of establishing the difference in the prenatal period. Other endocrine diseases, such as congenital hypothyroidism, metabolic alterations with facial dysmorphism, such as mucopolysaccharidosis (Hurler, Hunter, and Maroteaux-Lamy syndromes), besides gangliosidosis and Pompe disease, should be ruled out.1
ETIOPATHOGENESIS
BWS is caused by a variety of genetic and/or epigenetic alterations that usually affect the regulation of genes imprinted on chromosome 11p15.52,6 which may result in an heterogeneous clinical spectrum. Therefore, it is a paradigm of congenital abnormalities associated with genomic imprinting,1,5 a process that consists in a specific gene expression of parental origin. Up to 90% of cases are caused by an alteration in the expression of genes involved in cell cycle progression and somatic growth control, regulated by two independent imprinting centers (IC1 and IC2),1,6,7,22 which cover approximately 1 Mb.7
Across the entire genome, there are approximately 120 imprinted genes associated with 44 imprinting centers. In addition to BWS, other disorders have been well characterized, such as the Prader-Willi, Angelman, Temple, Kagami-Ogata, and Silver-Russell syndromes, together with transient neonatal diabetes mellitus and pseudohypoparathyroidism.7
Imprinting refers to the preferential or exclusive expression of the paternal or maternal allele of an imprinted gene. The imprinting gene expression is regulated by epigenetic mechanisms. The most common one is DNA methylation in the imprinting centers rich in CpG islands, which make up an important percentage of gene promoters.2 IC1 and IC2 are characterized by different methylation patterns of the maternal and paternal alleles,1 and, in normal conditions, IC1 of the paternal allele and IC2 of the maternal allele are methylated.5
The main cause is the loss of methylation in IC2 -in 50-60% of cases (Table 2)-, which is located centromeric to IC1.1,2,4,5,9,10,22 It results in a reduced expression of the cyclin dependent kinase inhibitor 1C (CDKN1C) gene,1,22 which works as a tumor suppressor gene and a negative regulator of fetal growth,2 normally expressed by the maternal chromosome.1,22 Individuals who carry this genetic alteration tend to develop macroglossia,14 hepatoblastoma, neuroblastoma, and adrenal tumors.12
Table 2. Etiopathogenic factors in Beckwith-Wiedemann syndrome, frequency and associated findings1,2,4,7,9,13,17,31,32
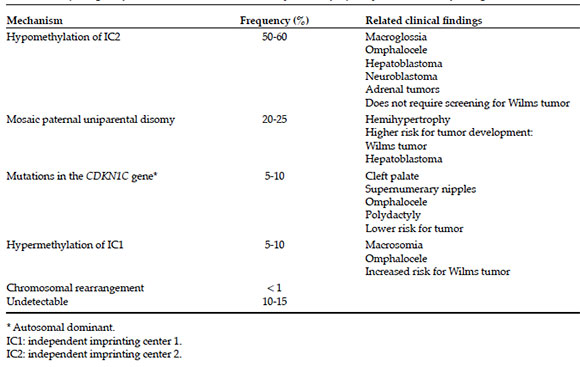
In addition, maternal point mutations in the CDKN1C gene account for 5-10% of cases1,4-6,9,10 and are responsible for 5% of sporadic cases1,4 and half of the cases with a positive family history,1,6,7 preferably maternal and with an autosomal dominant inheritance pattern.21 For this reason, most BWS cases are rare.10,21 Polydactyly, supernumerary nipples, and cleft palate are most commonly observed in this type of gene alteration.11
Omphalocele occurs more frequently in patients with hypomethylation of IC22,6 or point mutations in the CDKN1C gene.2,6,13 However, the risk for tumors is significantly lower in these two situations,2,6 and multiple studies have concluded that the loss of methylation of IC2 does not imply a higher risk and, therefore, does not require screening for Wilms tumor.2
In addition, 5-10% of cases are caused by hypermethylation of IC1,1,4,5,10,17 which results in the regulation of the biallelic expression of the insulin-like growth factor 2 (IGF2), normally expressed in the paternal allele, and the non-coding ribonucleic acid (RNA) of the oncosuppressor H19 gene, normally expressed in the maternal allele.1,2,4,22 The presence of macrosomia,4 omphalocele,13 and a higher risk for Wilms tumor is more commonly associated with this type of epigenetic alteration.4,13
The alteration in IC1 and IC2 methylation is explained by mosaic paternal uniparental disomy, which occurs in 20-25% of cases1,2,5,6,9,10 and is associated with additional phenotypic features,1 such as hemihypertrophy4,13 and a higher risk for tumor development, especially Wilms tumor2,6,12 and hepatoblastoma.13,23
In general, less than 1% of cases are caused by chromosomal rearrangements, such as duplications, translocations, inversions, deletions, which encompass IC group genes,1,5,22 and approximately 10-15% of clinically diagnosed individuals do not have a detectable molecular defect, even if they have an evident phenotype.1,2,5,6
TUMOR SURVEILLANCE PROTOCOLS
Although all these mechanisms play a role in the pathogenesis of BWS that remains unknown,6 the knowledge of (epi)genotype-phenotype correlations has prompted recommendations to propose different health care strategies, including tumor surveillance protocols based on molecular classification, aimed at standardizing clinical practice.5,11
In view of the genetic findings described above, an abdominal ultrasound should be done (every 3-4 months during childhood to rule out Wilms tumor6,23 and neuroblastoma). Other studies that may be part of this screening include an annual chest X-ray and an abdominal computed tomography in the case of nephromegaly or any suspected observation.16 Besides, serum alpha-fetoprotein levels (every 2-3 months in the first 4 years) should be measured to screen for hepatoblastoma.6,16,23 Likewise, chorionic gonadotropin and catecholamine values should be determined to detect germ cell tumors and neuroblastoma, respectively. A urinalysis should also be done annually as part of the assessment. An early detection may warrant an adequate management of high-risk malignancies.16 Screening for tumors should be encouraged, especially for those with paternal uniparental disomy.23
GENETIC COUNSELING
As discussed here, most cases of BWS are rare; therefore, the studied case is, in general, the only one affected in the family group. Except when a point mutation in the CDKN1C gene is observed, which shows an autosomal dominant inheritance pattern; those with this genetic alteration have a 50% risk for recurrence. The prognosis varies depending on the clinical presentation of this disease, which may be different even among family cases.
For this reason, follow-up and management of these patients should be done in an early and individual manner so as to minimize the complications that may arise and provide available treatment.
The objective of this article is to describe the current status of BWS, learn how to make a diagnosis based on clinical features, and rule out differential diagnoses. The etiopathogenic causes vary; it is necessary to understand them to guide interdisciplinary medical surveillance protocols, which should be customized to each patient and include timely family genetic counseling.
1. Mussa A, Russo S, De Crescenzo A, et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet 2016; 24(2):183-90. [ Links ]
2. Brzezinski J, Shuman C, Choufani S, et al. Wilms tumour in Beckwith-Wiedemann syndrome and loss of methylation at imprinting centre 2: revisiting tumour surveillance guidelines. Eur J Hum Genet 2017; 25(9):1031-9. [ Links ]
3. Giabicani É, Brioude F, Le Bouc Y, et al. Imprinted disorders and growth. Ann Endocrinol (Paris) 2017; 78(2):112-3. [ Links ]
4. Lin HY, Chuang CK, Tu RY, et al. Epigenotype, genotype, and phenotype analysis of patients in Taiwan with Beckwith-Wiedemann syndrome. Mol Genet Metab 2016; 119(1-2):8-13. [ Links ]
5. Luk HM. Clinical and molecular characterization of Beckwith-Wiedemann syndrome in a Chinese population. J Pediatr Endocrinol Metab 2017; 30(1):89-95. [ Links ]
6. Maas SM, Vansenne F, Kadouch DJ, et al. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am JMed Genet A 2016; 170(9):2248-60. [ Links ]
7. Tenorio J, Romanelli V, Martin-Trujillo A, et al. Clinical and molecular analyses of Beckwith-Wiedemann syndrome: Comparison between spontaneous conception and assisted reproduction techniques. Am J Med Genet A 2016; 170(10):2740-9. [ Links ]
8. Zammit M, Caruana E, Cassar D, et al. Beckwith-Wiedemann Syndrome Review: A Guide for the Neonatal Nurse. Neonatal Netw 2017; 36(3):129-33. [ Links ]
9. Inoue T, Nakamura A, Matsubara K, et al. Continuous hypomethylation of the KCNQ1OT1:TSS-DMR in monochorionic twins discordant for Beckwith-Wiedemann syndrome. Am J Med Genet A 2017; 173(10):2847-50. [ Links ]
10. Mussa A, Molinatto C, Cerrato F, et al. Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics 2017; 140(1):e20164311. [ Links ]
11. Mussa A, Di Candia S, Russo S, et al. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur J Med Genet 2016; 59(1):52-64. [ Links ]
12. Cheungpasitporn W, Erickson SB. Beckwith-Wiedemann syndrome and recurrent bilateral renal calculi. Urol Ann 2017; 9(1):113-4. [ Links ]
13. Eggermann K, Bliek J, Brioude F, et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. Eur J Hum Genet 2016; 24(10):1377-87. [ Links ]
14. Cielo CM, Duffy KA, Vyas A, et al. Obstructive sleep apnoea and the role of tongue reduction surgery in children with Beckwith-Wiedemann syndrome. Paediatr Respir Rev 2018; 25:58-63. [ Links ]
15. Maas SM, Kadouch DJ, Masselink AC, et al. Taste and speech following surgical tongue reduction in children with Beckwith-Wiedemann síndrome. J Craniomaxillofac Surg 2016; 44(6):659-63. [ Links ]
16. Lapunzina Badía P, del Campo Casanelles M, Delicado Navarro A, et al. Guía clínica para el seguimiento de pacientes con síndrome de Beckwith-Wiedemann. An Pediatr (Bare) 2006; 64(3):252-9. [ Links ]
17. Spivey PS, Bradshaw WT. Recognition and management of the infant with Beckwith-Wiedemann Syndrome. Adv Neonatal Care 2009; 9(6):279-84. [ Links ]
18. MacFarland SP, Mostoufi-Moab S, Zelley K, et al. Management of adrenal masses in patients with Beckwith-Wiedemann syndrome. Pediatr Blood Cancer 2017; 64(8):26432. [ Links ]
19. Caza T, Manwaring J, Riddell J. Recurrent, bilateral, and metastatic pheochromocytoma in a young patient with Beckwith-Wiedemann syndrome: A genetic link? Can Urol Assoc J 2017; 11(5):e240-3. [ Links ]
20. Callea M, Yavuz I, Clarich G, et al. A case of Beckwith-Wiedemann syndrome with peculiar dental findings. Eur J Paediatr Dent 2016; 17(4):315-7. [ Links ]
21. Ferianec V, Bartova M. Beckwith-Wiedemann syndrome with overlapping Perlman syndrome manifestation. J Matern Fetal Neonatal Med 2014; 27(15):1607-9. [ Links ]
22. Mussa A, Russo S, de Crescenzo A, et al. Fetal growth patterns in Beckwith-Wiedemann syndrome. Clin Genet 2016; 90(1):21-7. [ Links ]
23. Mussa A, Ferrero GB. Serum alpha-fetoprotein screening for hepatoblastoma in Beckwith-Wiedemann syndrome. Am J Med Genet A 2017; 173(3):585-7. [ Links ]

