










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.6 Buenos Aires Dec. 2018
http://dx.doi.org/10.5546/aap.2018.437
ACTUALIZACIÓN
http://dx.doi.org/10.5546/aap.2018.437
Principales entidades genéticas asociadas con dientes supernumerarios
Prof. Francisco Cammarata-Scalisia, Prof. Andrea Avendañoa y Dr. Michele Calledb
a. Unidad de Genética Médica, Departamento de Pediatría, Facultad de Medicina, Universidad de Los Andes. Venezuela.
b. Unidad de Odontología, Ospedale Pediatrico Bambino Gesù, IRCCS, Roma, Italia.
Correspondencia: Prof. Francisco Cammarata-Scalisi: francocammarata19@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 5-12-2017
Aceptado: 29-6-2018
RESUMEN
Los dientes supernumerarios representan una anomalía dental frecuente en los seres humanos. Esta afección se define como la presencia de una mayor cantidad de dientes que los previstos en cuanto a los dientes de leche o a los permanentes. Según se ha notificado, la prevalencia varía entre el 0,2 % y el 3 % y es más frecuente en los varones que en las mujeres. Su etiología es heterogénea, sumamente variable y, en la mayoría de los casos, idiopática. Sin embargo, la presencia de múltiples dientes supernumerarios retenidos o erupcionados es infrecuente y está asociada con ciertos síndromes genéticos, como displasia cleidocraneal, poliposis adenomatosa familiar, síndrome tricorrinofalángico de tipo I, síndrome de Rubinstein-Taybi, síndrome de Nance-Horan, síndrome de Opitz G/BBB, síndrome oculofaciocardiodental y síndrome de Robinow (autosómico dominante). Se deben considerar los dientes supernumerarios para diagnosticar estas entidades a fin de ofrecer un abordaje interdisciplinario, además de brindar asesoramiento genético familiar adecuado.
Palabras clave: Diente supernumerario; Anomalías dentales; Genética.
INTRODUCCIÓN
Los dientes supernumerarios (DS), o hiperdoncia, representan una anomalía dental frecuente en los seres humanos. 1-4 Esta afección se define como la presencia de una mayor cantidad de dientes que los presentes en la dentición de leche o permanente.4,5 Según se ha notificado, la prevalencia oscila entre el 0,2 % y el 3 %,4 varía según la población y es más frecuente en los varones que en las mujeres, con una proporción de aproximadamente 2:1, 4.1 Esta anomalía es etiológicamente heterogénea y sumamente variable,y difiere en términos de cantidad, ubicación, morfología, relación con otros dientes, presencia junto con los dientes de leche o problemas permanentes y asociados, como la retención.2,3 Otras complicaciones incluyen el fracaso de la dentición, la rotación o el desplazamiento de los dientes adyacentes, la dilaceración, la reabsorción radicular, el apiñamiento, la maloclusión, la formación de fístulas y quistes, y el desarrollo tardío o anormal de las raíces de los dientes permanentes.4
Más frecuentemente, los DS se ubican en el incisivo superior y se denominan mesiodens, y en general son cónicos, pequeños y con forma de clavija.1 Por otra parte, cuando están en las regiones molares adyacentes o distales, se denominan paramolares o distomolares, respectivamente.4
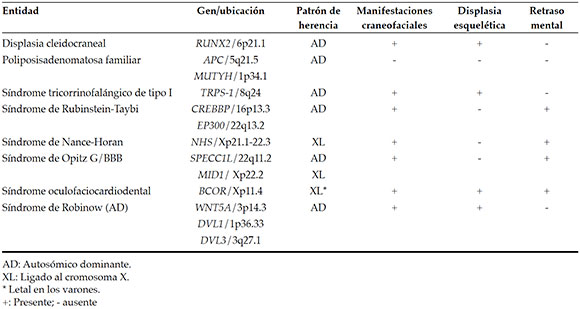
En la mayoría de los casos, los DS son idiopáticos,2,3 autosómicos dominantes y no sindrómicos (OMIM 108700) y se presentan con un patrón de transmisión autosómica recesiva o ligados al cromosoma X.1,4 Sin embargo, es rara la presencia de múltiples DS retenidos o erupcionados, y la mayoría de los casos están asociados con ciertos síndromes genéticos.2-5 Según la bibliografía, ocho entidades muy diferentes presentan los DS como característica distintiva, mencionadas en la Tabla 1 y desarrolladas en esta revisión. El objetivo de la misma es resumir las ocho entidades clínicas con diferentes patrones hereditarios cuyas características principales incluyen DS, para así ofrecer atención médica interdisciplinaria que incluya el examen dental y el asesoramiento genético oportuno.
Tabla 1. Entidades fuertemente asociadas con los dientes supernumerarios
DISPLASIA CLEIDOCRANEAL
La displasia cleidocraneal (OMIM 119600) es una displasia esquelética congénita rara, con un patrón de herencia autosómica dominante.6-11 Aproximadamente el 40 % de los pacientes parecen tener mutaciones espontáneas.La prevalencia es de 1 en 1 000 0008 y afecta a ambos sexos por igual.8,11 Su penetrancia es completa y su expresividad, ampliamente variable.8 La causa principal es una haploinsuficiencia en el gen RUNX2 (OMIM 600211), que se encuentra en el cromosoma 6p21.19-12 y codifica un factor de transcripción esencial para la diferenciación osteoblástica y el desarrollo esquelético.1
Los niños que sufren esta entidad se caracterizan por presentar baja estatura, cierre tardío de las fontanelas y las suturas craneales,1,9,11,13 aspecto aplanado del cráneo (braquicefalia),8,10 huesos wormianos,8,10,11 frente prominente, hipertelorismo,8,10 tabique nasal amplio y deprimido,8 hipoplasia facial media,8,9 paladar alto y estrecho,10,11 hendidura del paladar blando y del paladar duro,10 prognatismo mandibular,8 hipoplasia o aplasia clavicular(aspecto de hombros caídos),1,8-10,13 deformación de los omóplatos y del esternón, aplasia o costillas cervicales adicionales, tórax cónico (acampanado), cifoescoliosis,10 displasia congénita de la cadera,11 sínfisis púbica amplia y con cierre tardío,1,11 hiperlaxitud articular, laxitud muscular y desarrollo intelectual normal.10
La radiografía es la prueba más importante para confirmar el diagnóstico; en ella pueden observarse hipoplasia o aplasia clavicular, líneas de sutura amplias y fontanelas grandes, centros accesorios de osificación de los huesos de la cabeza, lo que da un aspecto de gran cantidad de huesos wormianos,áreas difusas de rarefacción con la mayoría de la osificación en los huesos de la frente y senos paranasales que están subdesarrollados y son estrechos.11
Las anomalías dentales varían ampliamente en cuanto a la gravedad,8 entre ellas los DS1,7,8,10,11,13 en ambas mandíbulas, con frecuencia en el área de los premolares; 11 a menudo, los dientes tienen forma aberrante debido al apiñamiento y a la retención.13 En los DS se retienen dientes de leche sin reabsorción radicular,7 que desplazan los dientes permanentes en desarrollo o no se observa su dentición,7,8,10,11,13 lo que produce múltiples dientes retenidos alrededor de los cuales surgen quistes dentígeros10,11 junto con una maloclusión grave.Se notificaron variaciones intrafamiliares en la cantidad de DS; de hecho, se propusieron factores ambientales y epigenéticos mediadores de estos fenotipos.14 A menudo, los dientes tienen esmalte hipoplásico y dilaceración radicular sin cemento celular. Algunos primordios o gérmenes dentarios se deforman y son rudimentarios, y también se observan microdoncia y geminación.7 Si bien la expresividad de la displasia cleidocraneal es variable, es posible realizar un diagnóstico temprano teniendo en cuenta las características dismórficas y las manifestaciones en la cavidad bucal.4
POLIPOSIS ADENOMATOSA FAMILIAR
La poliposis adenomatosa familiar (OMIM 175100) es una enfermedad que se caracteriza por la presencia de, al menos, 100 adenomas del intestino grueso y varias manifestaciones extracolónicas, con un patrón de herencia autosómica dominante.15,16 Se estima que la prevalencia es de aproximadamente 1en 10 000 nacidos vivos.17 Su causa son las mutaciones en la estirpe germinal en el gen APC (OMIM 611731)15-18 y, con menor frecuencia, las mutaciones en el gen MUTYH (OMIM 604933).15
El gen APC se encuentra en el cromosoma 5q21.5, que es un gen supresor tumoral que tiene una función central en la vía de señalización Wnt.2,17,18 La poliposis adenomatosa familiar es el resultado de una serie de cambios genéticos, que incluyen la activación de los oncogenes o la inactivación de los genes supresores tumorales, variaciones epigenéticas, además de otros cambios cromosómicos.19 Los factores ambientales y alimentarios pueden contribuir a la variación en la expresión clínica.16 Las mutaciones somáticas en el gen APC también son un evento molecular clave en el cáncer colorrectal esporádico presente en alrededor del 80 % de los pacientes.18
La cantidad y el tamaño de los adenomas aumentan drásticamente desde la pubertad hasta que el tubo digestivo se llena por completo de pólipos displásicos,16 con el desarrollo de cáncer colorrectal hacia los 40 años en casi el 100 % de los individuos.20 Entre los síntomas frecuentes se incluyen dolor abdominal, diarrea, rectorragia y melena. Los pacientes pueden tener deshidratación grave debido al desequilibrio electrolítico y la deshidratación a causa de la diarrea. Los adenomas en la ampolla de Vater pueden obstruir el flujo de la bilis y de las enzimas pancreatobiliares, lo que produce pancreatitis aguda.16 Entre las alteraciones extracolónicas, se incluyen otros tipos de cáncer, pólipos gástricos y duodenales, tumores desmoides,17 hipertrofia congénita del epitelio pigmentario retinal,17,21 osteomas,21 quistes epidérmicos17 y anomalías orales,21 que se notificaron en el 58-100 % de los individuos afectados.17 Entre estas, se incluyen los DS,2,17,22 que se observaron en el 11-27 % de los pacientes, mayormente entre los dientes del hueso alveolar o unidos al folículo de un diente retenido, y los sitios frecuentes son los dientes del sector anterior y alrededor de los caninos;2también se observan quistes dentígeros,22 dientes retenidos,17,22 ausencia congénita de uno o más dientes,22 odontomas17,22 y lesiones óseas en las mandíbulas.17
SÍNDROME TRICORRINOFALÁNGICO DE TIPO I
El síndrome tricorrinofalángico de tipo I (OMIM 190350) es un trastorno genético raro que se caracteriza por anomalías esqueléticas y craneofaciales distintivas, con un patrón de herencia autosómica dominante debido a defectos en el gen TRPS-1 (OMIM 604386), que se encuentra en el cromosoma 8q24; estos se identificaron como la causa más frecuente.23-25 El gen codifica el represor de la transcripción de la proteína con los dedos de cinc que participa en la regulación de la modulación del condrocito y en el desarrollo del pericondrio.2,25,26
Esta entidad se caracteriza por cabello fino, escaso y de crecimiento lento,23-26 línea de nacimiento del cabello alta,26 cejas gruesas en el medio y escasas en los extremos (signo de Hertoghe),24,25 orejas prominentes,25-27 hipoplasia facial media,27 rinofima,23,24,25 surco nasolabial plano y largo, labio superior delgado,25-27 protuberancia debajo del labio inferior,paladar ojival27,28 y anomalías dentales, como maloclusión28 y múltiples DS erupcionados.27-29 También se observan epífisis cónicas en las falanges de las manos,23-26 deformidades de las articulaciones interfalángicas que se asemejan a las de la artritis reumatoide,23 baja estatura y, con frecuencia —aunque no es un signo patognomónico—, displasia de cadera (enfermedad similar a la de Legg-Calve-Perthes en la cabeza femoral).24,25
Se describen tres subtipos junto con estas manifestaciones clínicas frecuentes. Además, los pacientes con síndrome tricorrinofalángico de tipo II o con síndrome de Langer-Giedion (OMIM 150230) presentan retraso mental, exostosis cartilaginosas múltiples y anomalías cutáneas. La presencia de braquidactilia grave y baja estatura severa con la ausencia de exostosis diferencia el síndrome tricorrinofalángico de tipo III o síndrome de Sugio-Kajii (OMIM 190351).24,25
SÍNDROME DE RUBINSTEIN-TAYBI
El síndrome de Rubinstein - Taybi (OMIM 180849 y 613684) es un trastorno autosómico dominante raro del neurodesarrollo, causado por mutaciones en el gen CREBBP (OMIM 600140) que se encuentra en el cromosoma 16p13.3 y codifica una proteína de unión a CREB, y en el gen EP300 (OMIM 602700), que se encuentra en el cromosoma 22q13.2 y codifica la proteína p300 asociada a E1A.30,31 Ambos genes actúan como coactivadores transcripcionales en la expresión de los genes involucrados en la embriología, la proliferación y la diferenciación celular y la supresión tumoral. También actúan como histona acetiltransferasas cruciales para la expresión génica;30,32 por lo tanto, este se considera un síndrome genético causado por una epigenética alterada.33 Se describieron aproximadamente 230 mutaciones causales en el gen CREBBP y 28 en el gen EP300, lo que representa aproximadamente del 50 % al 70 % y del 5 % al 8 % de los casos, respectivamente.30,31 Además, las características fenotípicas asociadas con las mutaciones en el gen EP300 son menos graves y bastante más variables que en los pacientes con mutaciones en el gen CREBBP.33 Esta entidad afecta a los varones y las mujeres por igual, con una prevalencia de 1 cada 100 000-125 000 nacidos vivos.31
Se caracteriza por un amplio rango de anomalías congénitas múltiples,como pulgares y dedos gordos anchos y dismorfismos craneofaciales30,31 (microcefalia,32,34 frente prominente,34 cejas arqueadas, pestañas largas,34,35 ptosis palpebral,34 fisuras palpebrales hacia abajo,30,31,34 pliegue del epicanto, obstrucción del conducto nasolagrimal, estrabismo,34 tabique nasal ancho, nariz en forma de pico,30,31,34,35 columela colgante,30,31 orejas ahuecadas o anguladas en la parte posterior,36 mohín o sonrisa inusual30,31,34 y micrognacia).34,35 El examen intraoral muestra paladar ojival, talón cuspídeo en los incisivos superiores de los dientes permanentes30,31,34,37,38 en más del 90 % de los casos,34 lo que resulta en caries, surcos dentales susceptibles e irritación de la lengua durante el habla y la masticación, hipodoncia,38 DS retenidos38,39 y dientes congénitos.34,38 Además, se observa deficiencia del crecimiento posnatal, discapacidad intelectual de moderada a grave30,31,35 y un leve aumento de la predisposición a tener cáncer.30,31
SÍNDROME DE NANCE-HORAN
El síndrome de Nance-Horan (OMIM 302350), o síndrome de catarata y defectos dentales, es un trastorno hereditario ligado al cromosoma X raro, causado por mutaciones en el gen NHS (OMIM 300457), que se encuentra en la región Xp21.1-22.340,41 y codifica tres isoformas diferentes (NHS A-C) como resultado del corte y el empalme alternativos. La isoforma A regula la remodelación de la actina y la morfología celular y se expresa particularmente en múltiples tejidos, incluido el cristalino, el cerebro, el mesénquima craneofacial y los primordios, sin correlaciones genotipo-fenotipo.41,42
Esta entidad se caracteriza por catarata bilateral congénita, anomalías dentales, dismorfismos faciales (tabique nasal ancho, rinofima, pabellones auriculares antevertidos y grandes y cara larga y estrecha)40,42-44 y dedos de las manos cortos.40,43 Además, se observa retraso del desarrollo, retraso mental variable,40,44 autismo inconsistente40 y algunos defectos cardíacos congénitos.43,45 Los varones afectados presentan catarata nuclear congénita densa y bilateral y otras características oftalmológicas, por ejemplo, microftalmia, microcórnea, nistagmo y estrabismo. Las anomalías dentales incluyen incisivos en forma de destornillador y brotes molaresen los dientes de leche y permanentes, y los DS en el incisivo superior se han destacado como los rasgos fenotípicos más importantes, junto con el diastema.40,43,44 Las mujeres heterocigotas presentan opacidad del cristalino centrada en las suturas en forma de Y posteriores.42 Estas manifestaciones clínicas más leves con expresividad variable son posiblemente el resultado de una inactivación desviada del cromosoma X.43,44
SÍNDROME DE OPITZ G/BBB
El síndrome de Opitz G/BBB es una afección rara genéticamente heterogénea, que tiene un patrón autosómico dominante (OMIM 145410) causado por una mutación en el gen SPECC1L (OMIM 614140), que se encuentra en el cromosoma 22q11.2,46,47 responsable de producir la proteína citospina-A, que interactúa con los elementos citoesqueléticos y la estabilización de los microtúbulos,48 o que se presenta vinculado al cromosoma X (OMIM 300000) y es causado por una mutación en el gen MID1 (OMIM 300552), que se encuentra en el cromosoma Xp22.246,49 y produce la proteína de la línea media-1, responsable de la unión a microtúbulos.48
Esta entidad se caracteriza por varias anomalías a lo largo de la línea media del cuerpo,46 por ejemplo, signos craneofaciales (craneosinostosis,49 frente prominente, pico de viuda,48 párpados caídos,49 hipertelorismo,46,48,49 nariz ancha y plana,48,50orificios nasales antevertidos,48 labio superior delgado,48,49 micrognacia,50 orejas prominentes y de implantación baja), anomalías intraorales (paladar ojival,48,49 labio leporino o fisura palatina,46,49,51 anquiloglosia,50,51 lengua geográfica,50 lengua bifida,50,51 frenillo lingual corto,51 agenesia dental,50 DS,50,51 úvula bifida),51 insuficiencia velofaríngea, defectos laringotraqueoesofágicos,46,48 cardiopatia congènita,46,48,49 anomalías renales,49 hipospadias,46,48,49 escroto bifido, criptorquidia48 y defectos anales.46,48,49Además, se observan retrasos variables del desarrollo,48 trastornos del aprendizaje,49 trastornos neuropsiquiátricos, síntomas que coinciden con el espectro autista,48 anomalías cerebrales,46,49 convulsiones, hipoacusia, problemas relevantes con la alimentación, inmunodeficiencia, hipocalciemia, deficiencia de la hormona del crecimiento, enfermedades autoinmunitarias y anomalías esquelèticas.49 Ambos patrones de herencia son clínicamente indiferenciables;48 sin embargo, en los varones con mutaciones en el gen MID1 se hallaron criptorquidia y anomalías anales con mayor frecuencia que en aquellos sin mutaciones.47
SÍNDROMEOCULOFACIOCARDIODENTAL
El síndrome oculofaciocardiodental (OMIM 300166) es un trastorno dominante raro, ligado al cromosoma X, observado en las mujeres heterocigotas y que es letal en los varones.52,53 Es causado por mutaciones heterocigotas en el gen BCOR (OMIM 300485), que codifica una proteína conocida como correpresor BCL6 y se encuentra en el cromosoma Xp11.4.52,54,55 Este gen se expresa predominantemente durante la embriogènesis temprana, y la proteína codificada BCOR funciona como correpresor transcripcional.53,55
Esta entidad se caracteriza por anomalías oculares (catarata congènita, microftalmia52-55,57,58 o microcórnea,55 glaucoma secundario),53,58 características craneofaciales (cara larga y estrecha, tabique nasal alto,53,55-58 punta nasal ancha,54,56-58 nariz bífida,52,56 surco nasolabial largo,54 deformidad de las orejas),56 paladar alto y estrecho,52,54 fisura palatina,52-54,56,58 manifestaciones
dentales (la radiculomegalia de los dientes permanentes es una característica sistemática,52-56 además de dentición tardía, dientes de leche persistentes,54,56,57 oligodoncia52,53 y DS).53,56Estas últimas anomalías se deben a que el gen BCOR se expresa en el epitelio y el mesènquima durante las primeras etapas del desarrollo de los dientes.53 Se observan anomalías cardíacas (comunicación interventricular, comunicación interauricular52-55,57 y prolapso de la válvula mitral)53,57 y anomalías esqueléticas (sindactiliadel segundo y tercer dedo del pie,54,55 dedos en martillo55 y sinostosis radiocubital).55,58 Además, se presentan retraso mental, trastorno por déficit de atención con hiperactividad e hipoacusia.58
SÍNDROME DE ROBINOW (AUTOSÓMICO DOMINANTE)
El síndrome de Robinow es un trastorno raro genéticamente heterogéneo, con un patrón autosómico dominante (OMIM 180700, 616331 y 616894), que puede ser causado por mutaciones de sentido alterado en el gen WNT5A, miembro 5A de la familia del sitio de integración MMTV de tipo Wingless (OMIM 164975),5960 que codifica una proteína secretada involucrada en la cascada de señalización no canónica independiente de p-catenina.61 Además, también es causado por mutaciones sin sentido en el gen DVL1 (OMIM 601365) y en el gen DVL3 (OMIM 601368) del extremo C de la proteína adaptadora Dishevelled.62 Esta entidad también se presenta con un patrón autosómico recesivo (OMIM 268310).59,60 Su prevalencia es de 1 en 500 000 nacidos vivos.62
Este síndrome es una displasia esquelética que se caracteriza por enanismo mesomélico,59-61,63 braquidactilia,63,64 clinodactilia,64 pulgares bífidos,63 características craneofaciales definidas como "cara fetal",59,61-63 frente ancha y alta,61 frente prominente,59,62 ojos prominentes,59,61 hipertelorismo,61-64 tabique nasal amplio y deprimido,59,61,62 nariz corta con orificios nasales antevertidos y punta ancha,61,63,64 hipoplasia facial media,61-64 boca ancha,63micrognacia, irregularidades dentales,62 hipertrofia gingival, disoclusión dental,63 DS (exclusivamente en la forma dominante),2,64 hernia umbilical,64 hipoplasia genital59-61,63 y capacidad intelectual normal.59,61
Otras entidades que podrían presentar dientes supernumerarios entre las manifestaciones clínicas
Ocasionalmente, otras entidades genéticas podrían presentar DS como las anomalías dentales: acondroplasia,65 síndrome de Ellis-van Creveld,2,4 síndrome de Kreiborg-Pakistani, síndrome de Apert,2 síndrome de Crouzon,2,66mucopolisacaridosis tipo IV (síndrome de Morquio)67 y de tipo VI (síndrome de Marateaux-Lamy),67,68 síndrome de Goldenhar,69síndrome de Noonan,70neurofibromatosis tipo 1,71 síndrome de Ehlers-Danlos,2 síndrome de Hallermann-Streiff,2 síndrome de Nicolaides-Baraitser,72 síndrome de Zimmermann-Laband,2 epidermólisis ampollosa distrófica,73amelogénesis imperfecta-nefrocalcinosis,74 enfermedad de Fabry2,4 y labio leporino y fisura palatina no sindrómicos,4,75 entre otras.
COMENTARIO Y CONCLUSIONES
Para el diagnóstico preciso de los DS y su tratamiento es necesario realizar una evaluación clínica y una pesquisa radiológica integral.4 En la mayoría de los pacientes, se observan complicaciones clínicas, que los odontopediatras y los pediatras que ven a menudo a estos niños podrían diagnosticar de manera temprana y para las cuales podría planificarse un tratamiento interdisciplinario a largo plazo más eficaz,76,77 que podría restaurar la función y la estética. Se debe establecer un protocolo de prevención relevante por medio del cual se brinden instrucciones para la limpieza y el mantenimiento de la salud bucal recomendada.76
Se describieron ocho entidades genéticas diferentes e infrecuentes desde el punto de vista clínico que tienen los DS entre sus signos representativos. Cinco de ellas presentan un patrón de herencia autosómica dominante, dos están ligadas al cromosoma X y la restante tiene ambos patrones de herencia que dependen de la heterogeneidad de locus. A fin de reconocer estas entidades desde la perspectiva clínica es necesario ofrecer atención médica interdisciplinaria con base en los hallazgos, incluido el examen dental, y luego establecer el patrón de herencia para brindar asesoramiento genético familiar oportuno.
1. Takahashi M, Hosomichi K, Yamaguchi T, et al. Whole-exome sequencing analysis of supernumerary teeth occurrence in Japanese individuals. Hum Genome Var 2017;4:16046. [ Links ]
2. Lubinsky M, Kantaputra PN.Syndromes with supernumerary teeth.Am JMed Genet A. 2016;170(10):2611-6. [ Links ]
3. de Souza Batista FR, Bonardi JP, Silva LF, et al. Supernumerary teeth in nonsyndrome patient. J Craniofac Surg 2017;28(2):583-4. [ Links ]
4. Subasioglu A, Savas S, Kucukyilmaz E, et al. Genetic background of supernumerary teeth. Eur J Dent 2015;9(1):153-8. [ Links ]
5. Tanwar R, Jaitly V, Sharma A, et al. Non-syndromic multiple supernumerary premolars: Clinicoradiographic report of five cases. J Dent Res Dent Clin Dent Prospects 2017;11(1):48-52. [ Links ]
6. Callea M, Fattori F, Bertini ES, et al. Estudio clínico y molecular en una familia con displasia cleidocraneal. Arch Argent Pediatr 2017;115(6):e440-4. [ Links ]
7. Callea M, Fattori F, Yavuz I, Bertini E. A new phenotypic variant in cleidocranial dysplasia (CCD) associated with mutation c.391C>T of the RUNX2 gene. BMJ Case Rep 2012;2012:bcr1220115422. [ Links ]
8. Rocha R, Zasso MB, Floriano G, et al. Orthodontic traction in a patient with cleidocranial dysplasia: 3 years of followup. Am J Orthod Dentofacial Orthop 2014;146(1):108-18. [ Links ]
9. Shibata A, Machida J, Yamaguchi S, et al. Characterisation of novel RUNX2 mutation with alanine tract expansion from Japanese cleidocranial dysplasia patient. Mutagenesis 2016;31(1):61-7. [ Links ]
10. Lewandowski B, Martula-Gala K, Brodowski R, Zych B. Multiple, supernumerary retained teeth in the course of cleido-cranial dysplasia. A case report. Dev Period Med 2015;19(4):503-7. [ Links ]
11. Singh S, Sharma S, Singh H, Wazir ND. Cleidocranial dysplasia: a case report illustrating diagnostic clinical and radiological findings. J ClinDiagn Res 2014;8(6):ZD19-20. [ Links ]
12. Callea M, Bellacchio E, Fattori F, et al. Acute myeloid leukemia in a 3 years old child with cleidocranial dysplasia. Leuk Lymphoma 2016;57(9):2189-91. [ Links ]
13. Atil F, Culhaoglu A, Kocyigit ID, et al. Oral rehabilitation with implant-supported fixed dental prostheses of a patient with cleidocranial dysplasia. J Prosthet Dent 2018;119(1):12-6. [ Links ]
14. Avendano A, Cammarata-Scalisi F, Rizal MF, et al. Cleidocranial dysplasia. A molecular and clinical review. Int Dent Res 2018;8(1):35-8. [ Links ]
15. Roncucci L, Pedroni M, Mariani F. Attenuated adenomatous polyposis of the large bowel: Present and future. World J Gastroenterol 2017;23(23):4135-9. [ Links ]
16. Waller A, Findeis S, Lee MJ. Familial adenomatous polyposis. J Pediatr Genet 2016;5(2):78-83. [ Links ]
17. Almeida FT, Pachêco-Pereira C, Porporatti AL, et al. Oral manifestations in patients with familial adenomatous polyposis: A systematic review and meta-analysis. J Gastroenterol Hepatol 2016;31(3):527-40. [ Links ]
18. Talseth-Palmer BA.The genetic basis of colonic adenomatous polyposis syndromes. Hered Cancer Clin Pract 2017;15:5. [ Links ]
19. Eshghifar N, Farrokhi N, Naji T, Zali M. Tumor suppressor genes in familial adenomatous polyposis. Gastroenterol Hepatol Bed Bench 2017;10(1):3-13. [ Links ]
20. Lami G, Galli A, Macri G, et al.Gastric and duodenal polyps in familial adenomatous polyposis patients: Conventional endoscopy vs virtual chromoendoscopy (fujinon intelligent color enhancement) in dysplasia evaluation. World J Clin Oncol 2017;8(2):168-77. [ Links ]
21. Li H, Zhang L, Jiang Q, et al. Identification a nonsense mutation of APC gene in Chinese patients with familial adenomatous polyposis. ExpTher Med 2017;13(4):1495-9. [ Links ]
22. Jasperson KW, Patel SG, Ahnen DJ.APC-Associated Polyposis Conditions. 1998 Dec 18 [Updated 2017 Feb 2]. In: Pagon RA, Adam MP, Ardinger HH, et al., (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. [Fecha de acceso: 2 de julio de 2018]. Disponibleen: https://www.ncbi.nlm.nih.gov/books/NBK1345/
23. Narayanan R, Chennareddy S.Crooked fingers and sparse hair: an interesting case of trichorhinophalangeal syndrome type 1. BMJ Case Rep 2015;2015: bcr2014207645. [ Links ]
24. Hufeland M, Rahner N, Krauspe R.Trichorhinophalangeal syndrome type I: a novel mutation and Perthes-like changes of the hip in a family with 4 cases over 3 generations. J Pediatr Orthop 2015;35(1):e1-5. [ Links ]
25. Merjaneh L, Parks JS, Muir AB, Fadoju D. A novel TRPS1 gene mutation causing trichorhinophalangeal syndrome with growth hormone responsive short stature: a case report and review of the literature. Int J PediatrEndocrinol 2014;2014(1):16. [ Links ]
26. Dias C, Isidoro L, Santos M, et al. Trichorhinophalangeal syndrome type I: A patient with two novel and different mutations in the TRPS1 gene. Case Rep Genet 2013;2013:748057. [ Links ]
27. Karacay S, Saygun I, Tunca Y, et al. Clinical and intraoral findings of a patient with tricho-rhino-phalangeal syndrome type I. J Indian Soc Pedod Prev Dent 2007;25(1):43-5. [ Links ]
28. Vaccaro M, Guarneri F, Barbuzza O, et al. A familial case of trichorhinophalangeal syndrome type I. PediatrDermatol 2009;26(2):171-5. [ Links ]
29. Candamourty R, Venkatachalam S, Karthikeyan B, Babu M.Trichorhinophalangeal syndrome type 1: A case report with literature review. J Nat Sci Biol Med 2012;3(2):209-11. [ Links ]
30. López M, Seidel V, Santibáñez P, et al. First case report of inherited Rubinstein-Taybi syndrome associated with a novel EP300 variant. BMC Med Genet 2016;17(1):97. [ Links ]
31. Spena S, Gervasini C, Milani D.Ultra-rare syndromes: The example of Rubinstein-Taybi syndrome. J PediatrGenet 2015;4(3):177-86. [ Links ]
32. Sellars EA, Sullivan BR, Schaefer GB.Whole exome sequencing reveals EP300 mutation in mildly affected female: expansion of the spectrum. Clin Case Rep 2016;4(7):696-8. [ Links ]
33. Masuda K, Akiyama K, Arakawa M, et al.Exome sequencing identification of EP300 mutation in a proband with coloboma and imperforate anus: Possible expansion of the phenotypic spectrum of Rubinstein-Taybi syndrome. Mol Syndromol 2015;6(2):99-103. [ Links ]
34. Stalin A, Varma BR, Jayanthi.Rubinstein-Taybi syndrome. J Indian Soc Pedod Prev Dent 2006;24 Suppl1:S27-30. [ Links ]
35. Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol 2016;25(4):135-45. [ Links ]
36. Solomon BD, Bodian DL, Khromykh A, et al. Expanding the phenotypic spectrum in EP300-related Rubinstein-Taybi syndrome. Am J Med Genet A 2015;167A(5):1111-6. [ Links ]
37. Deepthi DA, Shaheen VS, Kumar MH, et al. Broad thumb-hallux syndrome: diagnosis made on clinical findings. J Clin Diagn Res 2017;11(5):ZJ05-6. [ Links ]
38. Gunashekhar M, Hameed MS, Bokhari SK. Oral and dental manifestations in Rubinstein-Taybi syndrome: report of a rare case. Prim Dent Care 2012;19(1):35-8. [ Links ]
39. Tirali RE, Sar C, Cehreli SB. Oro-facio-dental findings of Rubinstein-Taybi syndrome as a useful diagnostic feature. J Clin Diagn Res 2014;8(1):276-8. [ Links ]
40. Gjorup H, Haubek D, Jacobsen P, Ostergaard JR. Nance-Horan syndrome-The oral perspective on a rare disease. Am J Med Genet A 2017;173(1):88-98. [ Links ]
41. Shoshany N, Avni I, Morad Y, et al. NHS gene mutations in Ashkenazi Jewish families with Nance-Horan syndrome. CurrEye Res 2017;42(9):1240-4. [ Links ]
42. Khan AO, Aldahmesh MA, Mohamed JY, Alkuraya F. Phenotype-genotype correlation in potential female carriers of X-linked developmental cataract (Nance-Horan syndrome). Ophthalmic Genet 2012;33(2):89-95. [ Links ]
43. Accogli A, Traverso M, Madia F, et al. A novel Xp22.13 microdeletion in Nance-Horan syndrome. Birth Defects Res 2017;109(11):866-8. [ Links ]
44. Tug E, Dilek NF, Javadiyan S, et al. A Turkish family with Nance-Horan syndrome due to a novel mutation. Gene 2013;525(1):141-5. [ Links ]
45. Chograni M, Rejeb I, Jemaa LB, et al. The first missense mutation of NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly. Eur J Hum Genet 2011;19(8):851-6. [ Links ]
46. Preiksaitiene E, Krasovskaja N, Utkus A, et al. R368X mutation in MIDI among recurrent mutations in patients with X-linked Opitz G/BBB syndrome. Clin Dysmorphol. 2015;24(1):7-12. [ Links ]
47. Kruszka P, Li D, Harr MH, et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. JMed Genet 2015;52(2):104-10. [ Links ]
48. Regan JP, Szymanski K, Podda S, et al. A surgical approach to the craniofacial defects of Opitz G/BBB syndrome. J Surg Case Rep 2017;2017(2):rjx032. [ Links ]
49. Giovani ÉM, Marinho KC,Andia-Merlin R.Dental treatment of a patient with Opitz G/BBB syndrome. Spec Care Dentist 2017;37(2):102-6. [ Links ]
50. da Silva Dalben G, Richieri-CostaA, deAssis taveira L.Tooth abnormalities and soft tissue alterations in patients with G/BBB syndrome.Oral Dis 2008;14(8):747-53. [ Links ]
51. Parashar SY, Anderson PJ, Cox TC, et al. Multidisciplinary management of Opitz G/BBB syndrome. Ann Plast Surg 2005;55(4):402-7. [ Links ]
52. O'Byrne JJ, Laffan E, Murray DJ, Reardon W. Oculo-facio-cardio-dental syndrome with craniosynostosis, temporal hypertrichosis, and deafness. Am J Med Genet A 2017;173(5):1374-7. [ Links ]
53. Kantaputra PN.BCOR mutations and unstoppable root growth: a commentary on oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells. J Hum Genet 2014;59(6):297-9. [ Links ]
54. Feberwee HE, Feenstra I, Oberoi S, et al. Novel BCOR mutations in patients with oculofaciocardiodental (OFCD) syndrome. Clin Genet 2014;85(2):194-7. [ Links ]
55. Kondo Y, Saitsu H, Miyamoto T, et al. A family of oculofaciocardiodental syndrome (OFCD) with a novel BCOR mutation and genomic rearrangements involving NHS. J Hum Genet 2012;57(3):197-201. [ Links ]
56. Sakaguchi K, Yagi T, Nagata J, et al. Patient with oculo-facio-cardio-dental syndrome treated with surgical orthodontics. Am J Orthod Dentofacial Orthop 2012;141(4 Suppl):S159-70. [ Links ]
57. Davoody A, Chen IP, Nanda R, et al. Oculofaciocardiodental syndrome: a rare case and review of the literature.Cleft Palate Craniofac J 2012;49(5):e55-60. [ Links ]
58. Di Stefano C, Lombardo B, Fabbricatore C, et al. Oculo-facio-cardio-dental (OFCD) syndrome: the first Italian case of BCOR and co-occurring OTC gene deletion. Gene 2015;559(2):203-6. [ Links ]
59. White JJ, Mazzeu JF, Hoischen A, et al. DVL3 alleles resulting in a -1 frameshift of the last exon mediate autosomal-dominant Robinow syndrome. Am J Hum Genet 2016;98(3):553-61. [ Links ]
60. Jeppesen BF, Hove HB, Kreiborg S, et al. Prenatal diagnosis of autosomal recessive Robinow syndrome using 3D ultrasound.Clin Case Rep 2017;5(7):1072-6. [ Links ]
61. White J, Mazzeu JF, Hoischen A, et al. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am J Hum Genet 2015;96(4):612-22. [ Links ]
62. Hosseini-Farahabadi S, Gignac SJ, Danescu A, et al. Abnormal WNT5Asignaling causes mandibular hypoplasia in Robinow syndrome. J Dent Res. 2017;96(11):1265-72. [ Links ]
63. Bunn KJ, Daniel P, Rösken HS, et al. Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. Am J Hum Genet 2015;96(4):623-30. [ Links ]
64. Mazzeu JF, Pardono E, Vianna-Morgante AM, et al. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am J Med Genet A 2007;143(4):320-5. [ Links ]
65. Raviraj J, Suman V, Suresh D, Kartik K. Achondroplasia with multiple supplemental supernumerary teeth and multiple talon cusps: A rare case report. Dent Res J (Isfahan)2017;14(3):219-22. [ Links ]
66. Torun GS, Akbulut A.Crouzon syndrome with multiple supernumerary teeth. Niger J Clin Pract 2017;20(2):261-3. [ Links ]
67. de Santana Sarmento DJ, de Carvalho SH, Melo SL, et al. Mucopolysaccharidosis: radiographic findings in a series of 16 cases. Oral Surg Oral Med Oral Pathol Oral Radiol 2015;120(6):e240-6. [ Links ]
68. Kantaputra PN, Kayserili H, GüvenY, etal. Oralmanifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis 2014;37(2):263-8. [ Links ]
69. Bogusiak K, Arkuszewski P, Skorek-Stachnik K, Kozakiewicz M. Treatment strategy in Goldenhar syndrome. J Craniofac Surg 2014;25(1):177-83. [ Links ]
70. Uloopi KS, Madhuri V, Gopal AS, et al. Multiple unerupted permanent teeth associated with Noonan syndrome. Ann Med Health Sci Res 2015;5(4):317-20. [ Links ]
71. Javed F, Ramalingam S,Ahmed HB, et al.Oral manifestations in patients with neurofibromatosis type-1 : a comprehensive literature review. Crit Rev Oncol Hematol 2014;91(2):123-9. [ Links ]
72. Al-Tamimi B, Abela S, Jeremiah HG, Evans R. Supernumeraries in Nicolaides-BaraitserSyndrome. Int J Paediatr Dent 2017;27(6):583-7. [ Links ]
73. Sharma S, Bedi S.Dystrophic epidermolysis bullosa associated with non-syndromic hypodontia. Indian Dermatol Online J 2013;4(4):296-9. [ Links ]
74. Kantaputra PN, Kaewgahya M, Khemaleelakul U, et al.Enamel-renal-gingival syndrome and FAM20A mutations. Am J Med Genet A 2014;164A(1):1-9. [ Links ]
75. Aspinall A, Raj S, Jugessur A, et al.Expanding the cleft phenotype: the dental characteristics of unaffected parents of Australian children with non-syndromic cleft lip and palate. Int J Paediatr Dent 2014;24(4):286-92. [ Links ]
76. Martins RB, de Souza RS, Giovani EM. Cleidocranial dysplasia: report of six clinical cases. Spec Care Dentist 2014;34(3):144-50. [ Links ]
77. Chalakkal P, Krishnan R, De Souza N, Da Costa G. A rare occurrence of supplementary maxillary lateral incisors and a detailed review on supernumerary teeth. J Oral Maxillofac Pathol 2018;22(1):149. [ Links ]

