










Servicios Personalizados
Revista

Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.116 no.6 Buenos Aires dic. 2018
http://dx.doi.org/10.5546/aap.2018.437
REVIEW
http://dx.doi.org/10.5546/aap.2018.eng.437
Main genetic entities associated with supernumerary teeth
Prof. Francisco Cammarata-Scalisia, Prof. Andrea Avendañoa and Dr. Michele Calledb
a. Unit of Medical Genetics, Department of Pediatrics, Faculty of Medicine, University of The Andes. Venezuela.
b. Unit of Dentistry, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy.
E-mail address: Prof. Francisco Cammarata-Scalisi: francocammarata19@gmail.com
Funding: None.
Conflict of interest: None.
Received: 5-12-2017
Accepted: 29-6-2018
ABSTRACT
Supernumerary teeth represent a common human dental anomaly, defined as presence of extra teeth-more than the normal number foreseen in primary or permanent dentition. The prevalence has been reported between 0.2 to 3%, and is more frequent in males than females. The etiology is heterogeneous, highly variable and most of the cases are idiopathic. However, the presence of multiple impacted or erupted supernumerary teeth is rare and associated with some genetic syndromes: cleidocranial displasia, familial adenomatous polyposis, trichorhinophalangeal syndrome type I, Rubinstein-Taybi syndrome, Nance-Horan syndrome, Opitz G/BBB syndrome, oculofaciocardiodental syndrome and Robinow syndrome (autosomal dominant). The supernumerary teeth should be considered in order to possibly diagnose these entities with the aim of offering an interdisciplinary management and treatment, as well as offer adequate family genetic counseling.
Key words: Supernumerary tooth; Tooth abnormalities; Genetic.
INTRODUCTION
Supernumerary teeth (ST) or hyperdontia represent a common human dental anomaly,1-4 defined as extra teeth-more than the normal number of teeth present primary or permanent dentition.4,5 The prevalence has been reported between 0.2 to 3%,4 varied among populations, and is more frequent in males than females, with a ratio of approximately 2:1.4.1 This anomaly is etiologically heterogeneous and is highly variable, differing in numbers, location, morphology, relationship with other teeth, presence in the primary dentition, and/or permanent and associated problems such as impaction.2,3 Other complications include eruption failure, rotation or displacement of the adjacent teeth, dilacerations, root resorption, crowding, malocclusion, fistulas and cystic formation, as well as delayed or abnormal root development of permanent teeth.4
The most common ST are located in the maxillary incisor and is referred as mesiodens, which are typically conical, small and peg-shaped.1 Moreover, in the molar regions adjacent or distal are referred to as paramolars or distomolars, respectively.4
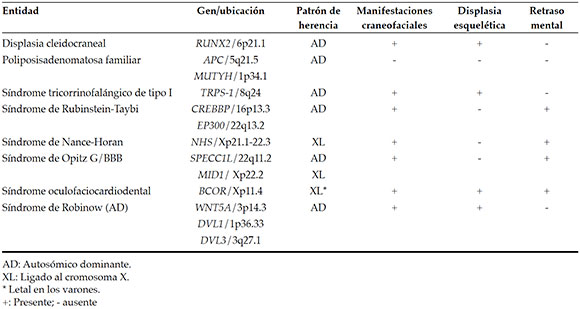
Most ST are idiopathic,2,3 autosomal dominant non-syndromic (OMIM 108700), autosomal recessive transmission pattern, or association with the X chromosom have been reported.1,4 However, the presence of multiple impacted or erupted ST is rare and mostly is associated with some genetic syndromes.2-5 According to what is found in the literature, eight very different entities present ST as distintive feature and these are mentioned in Table 1, and are developed in this review. The objective of this review is to summarize eight clinical entities with different inheritance patterns that present ST among their main findings, and thus provide interdisciplinary medical care that includes dental evaluation and provide timely genetic counseling.
Table 1. Entities with strong association to ST
CLEIDOCRANIAL DYSPLASIA
Cleidocranial displasia (OMIM 119600), is a rare congenital skeletal dysplasia with an autosomal dominant pattern of inheritance,6-11 Approximately 40% of patients appear to have spontaneous mutations. The prevalence is 1 in a million,8 and affect both sexes equally.8,11 Present complete penetrance and widely variable expressivity.8 The main cause is due by a haploinsufficiency in the RUNX2 gene (OMIM 600211), located on chromosome 6p21.1,9-12 and encodes an essential transcription factor for osteoblast differentiation and skeletal development.1
Is characterized by short stature, delayed closure of the cranial fontanelles and sutures,1,9,11,13 skull with flattened appearance (brachycephaly),810 wormian bones,8,10,11 frontal bossing, hypertelorism,8,10 wide and depressed nasal bridge,8 midfacial hipoplasia,8,9 high and narrow palate,10,11 cleft hard and soft palate,10 prognathic mandible,8 hypoplasia and/or aplasia of the clavicles (placement of the shoulders close to the front of the body),1,8-10,13 deformation of the scapulas and the sternum, aplasia or additional cervical ribs, the chest cone-shaped (a bell), kyphoscoliosis,10 congenital dislocation of hip,11 wide and delayed closure pubic symphysis,1,11 joint hypermobility, muscle laxity and normal intellectual development.10
The radiograph is the most important means by which the diagnosis can be confirmed, and shows hypoplastic or aplastic clavicle, broad suture lines and the fontannels are large, accessory centers of ossification bones of the head which gives the appearance of large numbers of wormian bones, diffuse areas of rarefaction with most ossification in the frontal bones and paranasal sinuses are usually underdeveloped and narrow.11
The dental abnormalities manifest a wide range of severity,8 including ST,1,7,8,10,11,13 localized teeth often have an aberrant shape related to impaction and crowding.13 ST retained deciduous teeth without root resorption,7 displace the developing permanent teeth and delayed or absent eruption,7,8,10,11,13 resulting in multiple impacted teeth around which dentigerous cysts frequently arise,10,11 and a serious malocclusion. Intrafamilial variations have been reported in the number of ST, indeed environmental and epigenetic factors have been proposed as the mechanism mediating these phenotypes.14 The teeth often have hypoplastic enamel and dilacerated roots without cellular cementum. Some tooth germs are deformed and rudimentary, and microdontia and twinning are also present.7 Although there is a variable expressivity of cleidocranial displasia early diagnosis through dysmorphic features and also oral findings are possible.4
FAMILIAL ADENOMATOUS POLYPOSIS
Familial adenomatous polyposis (OMIM 175100), is a disease characterized by the presence of at least 100 adenomas of the large bowel and several extracolonic manifestations with an autosomal dominant pattern of inheritance,15,16 and prevalence estimated to be approximately 1 in 10,000 live birts.17 It is caused by a germline mutations in the APC gene (OMIM 611731 ),15-18 and less frequently, by mutations in the MUTYH gene (OMIM 604933).15 tumour suppressor gene that plays a central role in the Wnt signalling pathway.2,1718 Familial adenomatous polyposis is the result of a series of genetic changes, including activation of oncogenes or inactivation of tumor suppressor genes, epigenetic variations in addition to chromosomal changes.19 Environmental and dietary factors can contribute to the variation in clinical expression.16 Somatic mutations in APC is also a key molecular event in sporadic colorectal cancer present in about 80% of patients.18
The number and size of adenomas increase drastically from puberty until the gastrointestinal tract becomes completely filled by dysplastic polyps,16 with the develop of colorectal cancer by the age of 40 years in nearly 100% of individuals.20 Common symptoms include abdominal pain, diarrhea, hematochezia, and melena. Patients may present with severe dehydration due to electrolyte imbalances and depletion from diarrea. Adenomas at the ampulla of Vater can obstruct bile flow and pancreaticobiliary enzymes, leading to acute pancreatitis.16 Among the extra colonic alterations, including other types of cancers, gastric and duodenal polyps, desmoids tumors,17 congenital hypertrophy of the retinal pigment epithelium,17,21 osteomas,21 epidermal cysts,17 and oral abnormalities,21 which are reported to be present from 58 to 100% of affected individuals.17 Among these include ST,2,17,22 appeared in 11-27% of patients, mostly between teeth in the alveolar bone or attached to follicle of an impacted tooth, common sites were anterior and around canines,2 dentigerous cysts,22 unerupted teeth,17,22 congenital absence of one or more teeth,22 odontomas,17,22 and osseous jaw lesions.17
TRICHORHINOPHALANGEAL SYNDROME TYPE I
Trichorhinophalangeal syndrome type I (OMIM 190350), is a rare genetic disorder characterized by distinctive craniofacial and skeletal abnormalities with an autosomal dominant pattern of inheritance due to defects in the TRPS-1 gene (OMIM 604386), located on chromosome 8q24, which have been identified as the most common cause,23-25 and encodes a zinc finger transcription repressor involved in the regulation of chondrocyte modulation and perichondrium development.2,25,26
Is characterised by fine, sparse and slow-growing scalp hair,23-26 high frontal hairline,26 medially thick and laterally sparse eyebrows (Herthoge sign),24,25 protruding ears,25-27 midfacial hypoplasia,27 bulbous pear-shaped nose,23-25 long and flat philtrum, thin upper lip,25-27 bulge under lower lip, high-arched palate,27,28 and dental anomalies such as malocclusion,28 and multiple erupted ST.27-29 Also cone-shaped phalangeal epiphyses of the hand,23-26 deformities of the interphalangeal joints resembling those in rheumatoid arthritis,23 short stature and frequently but not pathognomonic, hip displasia (Legg-Calve-Perthes like disease of the femoral head).24,25
Three subtypes have been described with these common clinical findings. Moreover, patients with trichorhinophalangeal syndrome type II or Langer-Giedion syndrome (OMIM 150230) have mental retradation, multiple cartilaginous exostoses and skin abnormalities. The presence of severe brachydactyly and severe short stature with the absence of exostoses differentiates trichorhinophalangeal syndrome type III or Sugio-Kajii syndrome (OMIM 190351).24,25
RUBINSTEIN-TAYBI SYNDROME
Rubinstein-Taybi syndrome (OMIM 180849 and 613684) is a rare autosomal dominant neurodevelopmental disorder, due to mutations in CREBBP gene (OMIM 600140), located on chromosome 16p13.3, encoding a CREB-binding protein, and EP300 gene (OMIM 602700), located on chromosome 22q13.2 and encodes E1A-associated protein p300.30,31 Both genes act as transcriptional coactivators in the expression of genes involved in embryology, cell growth and differentiation, and tumor suppression. They also act as a histone acetyltransferase crucial for gene expression,30,32 therefore, is regarded as a genetic syndrome caused by altered epigenetic.33 Approximately 230 causative mutations in CREBBP and 28 in EP300 have been described, accounting for approximately 50 to 70% and 5 to 8% of cases, respectively.30,31 Furthermore, the phenotypic features associated with EP300 mutations are less severe and quite variable that typical cases with CREBBP mutations.33 Affects males and females equally with a birth prevalence of 1 in 100,000-125,000 live birts.31
Is characterized by a wide range of multiple congenital anomalies as broad thumbs and halluces, craniofacial dysmorphisms,30,31 (microcefalia,32,34 prominent forehead,34 full and arched eyebrows, long eyelashes,34,35 ptosis of eyelids,34 downslanted palpebral fissures,30,31,34 epicanthal fold, nasal lacrimal duct obstruction, strabismus,34 broad nasal bridge, beaked nose,30,31,34,35 low hanging columella,30,31 cupped or posteriorly angulated ears,36 grimacing or unusual smile,30,31,34 and micrognathia).34,35 Intraoral examination which shows high arched palate, talons cusps on the upper incisors of the permanent dentition,30,31,34,37,38 have been observed in over 90% of cases,34 which resulting caries-susceptible developmental grooves and irritation of the tongue during speech and mastication, hypodontia,38 unerupted ST,38,39 and natal teeth.34,38 Moreover, postnatal growth deficiency, moderate to severe intellectual disability,30,31,35 and have a slightly increased predisposition to cancer.30,31
NANCE-HORAN SYNDROME
Nance-Horan syndrome (OMIM 302350) or cataract-dental syndrome is a rare X-linked hereditary disorder due to mutations in NHS gene (OMIM 300457), located within the region Xp21.1-22.3,40,41 and encodes three different isoforms (NHS A-C) as a result of alternative splicing. Isoform A is a regulator of actin remodeling and cell morphology, is particularly expressed in multiple tissues including the lens, brain, craniofacial mesenchyme and dental primordia, with no clear phenotype-genotype correlations.41,42
Is characterized by congenital bilateral cataract, dental abnormalities, facial dysmorphisms (broad nasal bridge, bulbous nose, large anteverted pinnae and long-narrow face),40,42-44 and short fingers.40,43 Furthermore, developmental delay, variable mental retardation,40,44 autism is more inconsistent,40 and some present congenital cardiac defects.43,45 Affected males have severe bilateral congenital dense nuclear cataracts and others ophthalmological features, including microphthalmia, microcornea, nystagmus, and strabismus. Dental abnormalities consist of screwdriver-shaped incisors and bud molars in both the primary and the permanent dentition, ST maxillary incisors have been emphasized as major phenotypic traits and diastema.40,43,44 Heterozygous females present lens opacities centered on the posterior Y-sutures.42 This milder clinical manifestations with variable expressivity are result of posible skewed X chromosome nactivation.43,44
OPITZ G/BBB SYNDROME
Opitz G/BBB syndrome is a rare genetically heterogeneous condition, with both an autosomal dominant form (OMIM 145410) which is caused by mutation in SPECC1L gene (OMIM 614140) located on chromosome 22q11.2,46,47 responsible for the production of cytospin-A which interacts with cytoskeletal elements and microtubule stabilization,48 and X-linked form (OMIM 300000) caused by mutation in MIDI gene (OMIM 300552) located on chromosome Xp22.2,46,49 and produces the midline-1 protein responsible for microtubule binding.48
Is characterized by several abnormalities along the midline of the body,46 as craniofacial findings (craniosynostosis,49 prominent forehead, widow's peak,48 hooded eyelids,49 hypertelorism,46,48,49 broad and flat nose,48,50 anteverted nares,48 thin upper lip,48,49 micrognathia,50 low-set prominent ears), intraoral anomalies (high-arched palate,48,49 cleft lip and/or palate,46,49,51 ankyloglossia,50,51 geographic tongue,50 bifid tongue,50,51 short lingual frenulum,51 tooth agenesis,50 ST,50,51 bifid uvula),51 velopharyngeal incompetence, laryngotracheal-oesophageal defects,46,48 congenital heart disease,46,48,49 renal anomalies,49 hypospadias,46,48,49
bifid scrotum, cryptorchidism,48 and anal defects.46,48,49 Furthermore, variable developmental delays,48 learning disabilities,49 neuropsychiatric disorders, symptoms consistent within the autism spectrum,48 brain anomalies,46,49 seizures, hearing loss, significant feeding problems, immune deficiency, hypocalcemia, growth hormone deficiency, autoimmune diseases and skeletal abnormalities.49 The two inheritance pattern are clinically indistinguishable,48 however, hypospadias and anal anomalies were found more commonly in male patients with MID1 mutations than in those without.47
OCULOFACIOCARDIODENTAL
SYNDROME
Oculofaciocardiodental syndrome (OMIM 300166) is a rare X linked dominant disorder, been seen in heterozygous females and lethal on males.52,53 Is caused by heterozygous mutations in the BCL-6 interacting corepressor, BCOR gene (OMIM 300485), located on chromosome Xp11.452,54,55 This gene is ubiquitously expressed during early embryogenesis and the encoded BCOR protein functions as a transcriptional corepressor.53,55
Is characterized by ocular abnormalities (congenital cataract, microphthalmia,52-55,57,58 and/or microcornea,55 secondary glaucoma),53,58 craniofacial feautures (long and narrow face, high-nasal bridge,53,55-58 broad nasal tip,54,56-58 bifid nose,52,56 long philtrum,54 ear deformity),56 high and narrow palate,52,54 cleft palate,52-54,56,58 dental findings (radiculomegaly of permanent teeth as consistent feature,52-56 delayed eruption and persistent primary dentition,54,56,57 oligodontia,52,53 and ST).53,56 These last anomalies are due to the BCOR gene is expressed in both dental epithelium and the mesenchyme during the early stages of tooth development.53 The cardiac anomalies (atrial and ventricular septal defects,52-55,57 and mitral valve prolapse),53,57 skeletal abnormalities (syndactyly of the second and third toes,54,55 hammer toes,55 and radioulnar synostosis).55,58 Furthermore, mental retardation, attention deficit/hyperactivity disorder, and hearing impairment.58
ROBINOW SYNDROME (AUTOSOMAL DOMINANT)
Robinow syndrome is a rare genetically heterogeneous disorder, with autosomaldominant form (OMIM 180700, 616331 and 616894), which can be caused by missense mutations in the secreted protein Wingless-type MMTV integration site family, member 5A, WNT5A gene (OMIM 164975),59,60 which encodes a protein that participates in the non-canonical, p-catenin-independent signaling cascade.61 Moreover, nonsense mutations in the C-terminal of the adapter protein Dishevelled DVL1 gene (OMIM 601365) and DVL3 gene (OMIM 601368).62 Also has a autosomal-recessive form (OMIM 268310).59,60 Present a prevalence of 1 in 500,000 live birts.62
Is a skeletal displasia characterized by mesomelic dwarfism,59-61,63 brachydactyly,63,64 clinodactyly,64 bifid thumbs,63 distintive craniofacial features characterized as "fetal face",59,61-63 high and broad forehead,61 frontal bossing,59,62 prominent eyes,59,61 hypertelorism,61-64 wide and depressed nasal bridge,59,61,62 short nose with anteverted nares and a broad tip,61,63,64 midfacial hypoplasia,61-64 broad mouth,63 micrognathia, dental irregularities,62 gingival hypertrophy, dental malocclusion,63 ST (exclusively in dominant form),2,64 umbilical hernia,64 genital hipoplasia,59-61,63 and normal intellect.59,61
OTHER ENTITIES THAT MAY HAVE ST AMONG THEIR CLINICAL FINDINGS
Eventually, other genetic entities may present ST as dental anomalies: achondroplasia,65 Ellis-van Creveld syndrome,2,4 Kreiborg-Pakistani syndrome, Apert syndrome,2 Crouzon syndrome,2,66 mucopolysaccharidoses type IV (Morquio syndrome),67 and type VI (Marateaux-Lamy syndrome),67,68 Goldenhar syndrome,69 Noonan syndrome,70 neurofibromatosis type-1,71 Ehlers-Danlos syndrome, Hallermann-Streiff syndrome,2 Nicolaides-Baraitser syndrome,72 Zimmermann-Laband syndrome,2 dystrophic epidermolysis bullosa,73 enamel-renal-gingival syndrome,74 Fabry disease,2,4 and non-syndromic cleft lip and palate,4,75 among others.
CONCLUSIONS
A precise diagnosis of ST and its management requires clinical examination and a comprehensive radiographic screening.4 The majority cause clinical complications, which Pediatric Dentists and Pediatricians who usually visit children, can contribute to early diagnosis and planning more effective long-term interdisciplinary treatment,76,77 that can restore function and aesthetics. Important role prevention protocol must be established where they were instructed about the cleaning and maintenance of desirable conditions of oral health.76
Eight different and infrequent genetic entities were described from the clinical point of view, which present ST as a representative finding. Five of them with an autosomal dominant inheritance pattern, two linked to the X chromosome and one with both inheritance patterns depending on the locus heterogeneity, Table 1. To recognize from the clinical point of view these entities is necessary to provide an interdisciplinary medical assistance according to the found findings and that includes the dental evaluation, and finally through the inheritance pattern to establish an opportune genetic family counseling.
1. Takahashi M, Hosomichi K, Yamaguchi T, et al. Whole-exome sequencing analysis of supernumerary teeth occurrence in Japanese individuals. Hum Genome Var 2017;4:16046. [ Links ]
2. Lubinsky M, Kantaputra PN.Syndromes with supernumerary teeth.Am JMed Genet A. 2016;170(10):2611-6. [ Links ]
3. de Souza Batista FR, Bonardi JP, Silva LF, et al. Supernumerary teeth in nonsyndrome patient. J Craniofac Surg 2017;28(2):583-4. [ Links ]
4. Subasioglu A, Savas S, Kucukyilmaz E, et al. Genetic background of supernumerary teeth. Eur J Dent 2015;9(1):153-8. [ Links ]
5. Tanwar R, Jaitly V, Sharma A, et al. Non-syndromic multiple supernumerary premolars: Clinicoradiographic report of five cases. J Dent Res Dent Clin Dent Prospects 2017;11(1):48-52. [ Links ]
6. Callea M, Fattori F, Bertini ES, et al. Estudio clínico y molecular en una familia con displasia cleidocraneal. Arch Argent Pediatr 2017;115(6):e440-4. [ Links ]
7. Callea M, Fattori F, Yavuz I, Bertini E. A new phenotypic variant in cleidocranial dysplasia (CCD) associated with mutation c.391C>T of the RUNX2 gene. BMJ Case Rep 2012;2012:bcr1220115422. [ Links ]
8. Rocha R, Zasso MB, Floriano G, et al. Orthodontic traction in a patient with cleidocranial dysplasia: 3 years of followup. Am J Orthod Dentofacial Orthop 2014;146(1):108-18. [ Links ]
9. Shibata A, Machida J, Yamaguchi S, et al. Characterisation of novel RUNX2 mutation with alanine tract expansion from Japanese cleidocranial dysplasia patient. Mutagenesis 2016;31(1):61-7. [ Links ]
10. Lewandowski B, Martula-Gala K, Brodowski R, Zych B. Multiple, supernumerary retained teeth in the course of cleido-cranial dysplasia. A case report. Dev Period Med 2015;19(4):503-7. [ Links ]
11. Singh S, Sharma S, Singh H, Wazir ND. Cleidocranial dysplasia: a case report illustrating diagnostic clinical and radiological findings. J ClinDiagn Res 2014;8(6):ZD19-20. [ Links ]
12. Callea M, Bellacchio E, Fattori F, et al. Acute myeloid leukemia in a 3 years old child with cleidocranial dysplasia. Leuk Lymphoma 2016;57(9):2189-91. [ Links ]
13. Atil F, Culhaoglu A, Kocyigit ID, et al. Oral rehabilitation with implant-supported fixed dental prostheses of a patient with cleidocranial dysplasia. J Prosthet Dent 2018;119(1):12-6. [ Links ]
14. Avendano A, Cammarata-Scalisi F, Rizal MF, et al. Cleidocranial dysplasia. A molecular and clinical review. Int Dent Res 2018;8(1):35-8. [ Links ]
15. Roncucci L, Pedroni M, Mariani F. Attenuated adenomatous polyposis of the large bowel: Present and future. World J Gastroenterol 2017;23(23):4135-9. [ Links ]
16. Waller A, Findeis S, Lee MJ. Familial adenomatous polyposis. J Pediatr Genet 2016;5(2):78-83. [ Links ]
17. Almeida FT, Pachêco-Pereira C, Porporatti AL, et al. Oral manifestations in patients with familial adenomatous polyposis: A systematic review and meta-analysis. J Gastroenterol Hepatol 2016;31(3):527-40. [ Links ]
18. Talseth-Palmer BA.The genetic basis of colonic adenomatous polyposis syndromes. Hered Cancer Clin Pract 2017;15:5. [ Links ]
19. Eshghifar N, Farrokhi N, Naji T, Zali M. Tumor suppressor genes in familial adenomatous polyposis. Gastroenterol Hepatol Bed Bench 2017;10(1):3-13. [ Links ]
20. Lami G, Galli A, Macri G, et al.Gastric and duodenal polyps in familial adenomatous polyposis patients: Conventional endoscopy vs virtual chromoendoscopy (fujinon intelligent color enhancement) in dysplasia evaluation. World J Clin Oncol 2017;8(2):168-77. [ Links ]
21. Li H, Zhang L, Jiang Q, et al. Identification a nonsense mutation of APC gene in Chinese patients with familial adenomatous polyposis. ExpTher Med 2017;13(4):1495-9. [ Links ]
22. Jasperson KW, Patel SG, Ahnen DJ.APC-Associated Polyposis Conditions. 1998 Dec 18 [Updated 2017 Feb 2]. In: Pagon RA, Adam MP, Ardinger HH, et al., (eds). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2017. [Fecha de acceso: 2 de julio de 2018]. Disponibleen: https://www.ncbi.nlm.nih.gov/books/NBK1345/
23. Narayanan R, Chennareddy S.Crooked fingers and sparse hair: an interesting case of trichorhinophalangeal syndrome type 1. BMJ Case Rep 2015;2015: bcr2014207645. [ Links ]
24. Hufeland M, Rahner N, Krauspe R.Trichorhinophalangeal syndrome type I: a novel mutation and Perthes-like changes of the hip in a family with 4 cases over 3 generations. J Pediatr Orthop 2015;35(1):e1-5. [ Links ]
25. Merjaneh L, Parks JS, Muir AB, Fadoju D. A novel TRPS1 gene mutation causing trichorhinophalangeal syndrome with growth hormone responsive short stature: a case report and review of the literature. Int J PediatrEndocrinol 2014;2014(1):16. [ Links ]
26. Dias C, Isidoro L, Santos M, et al. Trichorhinophalangeal syndrome type I: A patient with two novel and different mutations in the TRPS1 gene. Case Rep Genet 2013;2013:748057. [ Links ]
27. Karacay S, Saygun I, Tunca Y, et al. Clinical and intraoral findings of a patient with tricho-rhino-phalangeal syndrome type I. J Indian Soc Pedod Prev Dent 2007;25(1):43-5. [ Links ]
28. Vaccaro M, Guarneri F, Barbuzza O, et al. A familial case of trichorhinophalangeal syndrome type I. PediatrDermatol 2009;26(2):171-5. [ Links ]
29. Candamourty R, Venkatachalam S, Karthikeyan B, Babu M.Trichorhinophalangeal syndrome type 1: A case report with literature review. J Nat Sci Biol Med 2012;3(2):209-11. [ Links ]
30. López M, Seidel V, Santibáñez P, et al. First case report of inherited Rubinstein-Taybi syndrome associated with a novel EP300 variant. BMC Med Genet 2016;17(1):97. [ Links ]
31. Spena S, Gervasini C, Milani D.Ultra-rare syndromes: The example of Rubinstein-Taybi syndrome. J PediatrGenet 2015;4(3):177-86. [ Links ]
32. Sellars EA, Sullivan BR, Schaefer GB.Whole exome sequencing reveals EP300 mutation in mildly affected female: expansion of the spectrum. Clin Case Rep 2016;4(7):696-8. [ Links ]
33. Masuda K, Akiyama K, Arakawa M, et al.Exome sequencing identification of EP300 mutation in a proband with coloboma and imperforate anus: Possible expansion of the phenotypic spectrum of Rubinstein-Taybi syndrome. Mol Syndromol 2015;6(2):99-103. [ Links ]
34. Stalin A, Varma BR, Jayanthi.Rubinstein-Taybi syndrome. J Indian Soc Pedod Prev Dent 2006;24 Suppl1:S27-30. [ Links ]
35. Hamilton MJ, Newbury-Ecob R, Holder-Espinasse M, et al. Rubinstein-Taybi syndrome type 2: report of nine new cases that extend the phenotypic and genotypic spectrum. Clin Dysmorphol 2016;25(4):135-45. [ Links ]
36. Solomon BD, Bodian DL, Khromykh A, et al. Expanding the phenotypic spectrum in EP300-related Rubinstein-Taybi syndrome. Am J Med Genet A 2015;167A(5):1111-6. [ Links ]
37. Deepthi DA, Shaheen VS, Kumar MH, et al. Broad thumb-hallux syndrome: diagnosis made on clinical findings. J Clin Diagn Res 2017;11(5):ZJ05-6. [ Links ]
38. Gunashekhar M, Hameed MS, Bokhari SK. Oral and dental manifestations in Rubinstein-Taybi syndrome: report of a rare case. Prim Dent Care 2012;19(1):35-8. [ Links ]
39. Tirali RE, Sar C, Cehreli SB. Oro-facio-dental findings of Rubinstein-Taybi syndrome as a useful diagnostic feature. J Clin Diagn Res 2014;8(1):276-8. [ Links ]
40. Gjorup H, Haubek D, Jacobsen P, Ostergaard JR. Nance-Horan syndrome-The oral perspective on a rare disease. Am J Med Genet A 2017;173(1):88-98. [ Links ]
41. Shoshany N, Avni I, Morad Y, et al. NHS gene mutations in Ashkenazi Jewish families with Nance-Horan syndrome. CurrEye Res 2017;42(9):1240-4. [ Links ]
42. Khan AO, Aldahmesh MA, Mohamed JY, Alkuraya F. Phenotype-genotype correlation in potential female carriers of X-linked developmental cataract (Nance-Horan syndrome). Ophthalmic Genet 2012;33(2):89-95. [ Links ]
43. Accogli A, Traverso M, Madia F, et al. A novel Xp22.13 microdeletion in Nance-Horan syndrome. Birth Defects Res 2017;109(11):866-8. [ Links ]
44. Tug E, Dilek NF, Javadiyan S, et al. A Turkish family with Nance-Horan syndrome due to a novel mutation. Gene 2013;525(1):141-5. [ Links ]
45. Chograni M, Rejeb I, Jemaa LB, et al. The first missense mutation of NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly. Eur J Hum Genet 2011;19(8):851-6. [ Links ]
46. Preiksaitiene E, Krasovskaja N, Utkus A, et al. R368X mutation in MIDI among recurrent mutations in patients with X-linked Opitz G/BBB syndrome. Clin Dysmorphol. 2015;24(1):7-12. [ Links ]
47. Kruszka P, Li D, Harr MH, et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. JMed Genet 2015;52(2):104-10. [ Links ]
48. Regan JP, Szymanski K, Podda S, et al. A surgical approach to the craniofacial defects of Opitz G/BBB syndrome. J Surg Case Rep 2017;2017(2):rjx032. [ Links ]
49. Giovani ÉM, Marinho KC,Andia-Merlin R.Dental treatment of a patient with Opitz G/BBB syndrome. Spec Care Dentist 2017;37(2):102-6. [ Links ]
50. da Silva Dalben G, Richieri-CostaA, deAssis taveira L.Tooth abnormalities and soft tissue alterations in patients with G/BBB syndrome.Oral Dis 2008;14(8):747-53. [ Links ]
51. Parashar SY, Anderson PJ, Cox TC, et al. Multidisciplinary management of Opitz G/BBB syndrome. Ann Plast Surg 2005;55(4):402-7. [ Links ]
52. O'Byrne JJ, Laffan E, Murray DJ, Reardon W. Oculo-facio-cardio-dental syndrome with craniosynostosis, temporal hypertrichosis, and deafness. Am J Med Genet A 2017;173(5):1374-7. [ Links ]
53. Kantaputra PN.BCOR mutations and unstoppable root growth: a commentary on oculofaciocardiodental syndrome: novel BCOR mutations and expression in dental cells. J Hum Genet 2014;59(6):297-9. [ Links ]
54. Feberwee HE, Feenstra I, Oberoi S, et al. Novel BCOR mutations in patients with oculofaciocardiodental (OFCD) syndrome. Clin Genet 2014;85(2):194-7. [ Links ]
55. Kondo Y, Saitsu H, Miyamoto T, et al. A family of oculofaciocardiodental syndrome (OFCD) with a novel BCOR mutation and genomic rearrangements involving NHS. J Hum Genet 2012;57(3):197-201. [ Links ]
56. Sakaguchi K, Yagi T, Nagata J, et al. Patient with oculo-facio-cardio-dental syndrome treated with surgical orthodontics. Am J Orthod Dentofacial Orthop 2012;141(4 Suppl):S159-70. [ Links ]
57. Davoody A, Chen IP, Nanda R, et al. Oculofaciocardiodental syndrome: a rare case and review of the literature.Cleft Palate Craniofac J 2012;49(5):e55-60. [ Links ]
58. Di Stefano C, Lombardo B, Fabbricatore C, et al. Oculo-facio-cardio-dental (OFCD) syndrome: the first Italian case of BCOR and co-occurring OTC gene deletion. Gene 2015;559(2):203-6. [ Links ]
59. White JJ, Mazzeu JF, Hoischen A, et al. DVL3 alleles resulting in a -1 frameshift of the last exon mediate autosomal-dominant Robinow syndrome. Am J Hum Genet 2016;98(3):553-61. [ Links ]
60. Jeppesen BF, Hove HB, Kreiborg S, et al. Prenatal diagnosis of autosomal recessive Robinow syndrome using 3D ultrasound.Clin Case Rep 2017;5(7):1072-6. [ Links ]
61. White J, Mazzeu JF, Hoischen A, et al. DVL1 frameshift mutations clustering in the penultimate exon cause autosomal-dominant Robinow syndrome. Am J Hum Genet 2015;96(4):612-22. [ Links ]
62. Hosseini-Farahabadi S, Gignac SJ, Danescu A, et al. Abnormal WNT5Asignaling causes mandibular hypoplasia in Robinow syndrome. J Dent Res. 2017;96(11):1265-72. [ Links ]
63. Bunn KJ, Daniel P, Rösken HS, et al. Mutations in DVL1 cause an osteosclerotic form of Robinow syndrome. Am J Hum Genet 2015;96(4):623-30. [ Links ]
64. Mazzeu JF, Pardono E, Vianna-Morgante AM, et al. Clinical characterization of autosomal dominant and recessive variants of Robinow syndrome. Am J Med Genet A 2007;143(4):320-5. [ Links ]
65. Raviraj J, Suman V, Suresh D, Kartik K. Achondroplasia with multiple supplemental supernumerary teeth and multiple talon cusps: A rare case report. Dent Res J (Isfahan)2017;14(3):219-22. [ Links ]
66. Torun GS, Akbulut A.Crouzon syndrome with multiple supernumerary teeth. Niger J Clin Pract 2017;20(2):261-3. [ Links ]
67. de Santana Sarmento DJ, de Carvalho SH, Melo SL, et al. Mucopolysaccharidosis: radiographic findings in a series of 16 cases. Oral Surg Oral Med Oral Pathol Oral Radiol 2015;120(6):e240-6. [ Links ]
68. Kantaputra PN, Kayserili H, GüvenY, etal. Oralmanifestations of 17 patients affected with mucopolysaccharidosis type VI. J Inherit Metab Dis 2014;37(2):263-8. [ Links ]
69. Bogusiak K, Arkuszewski P, Skorek-Stachnik K, Kozakiewicz M. Treatment strategy in Goldenhar syndrome. J Craniofac Surg 2014;25(1):177-83. [ Links ]
70. Uloopi KS, Madhuri V, Gopal AS, et al. Multiple unerupted permanent teeth associated with Noonan syndrome. Ann Med Health Sci Res 2015;5(4):317-20. [ Links ]
71. Javed F, Ramalingam S,Ahmed HB, et al.Oral manifestations in patients with neurofibromatosis type-1 : a comprehensive literature review. Crit Rev Oncol Hematol 2014;91(2):123-9. [ Links ]
72. Al-Tamimi B, Abela S, Jeremiah HG, Evans R. Supernumeraries in Nicolaides-BaraitserSyndrome. Int J Paediatr Dent 2017;27(6):583-7. [ Links ]
73. Sharma S, Bedi S.Dystrophic epidermolysis bullosa associated with non-syndromic hypodontia. Indian Dermatol Online J 2013;4(4):296-9. [ Links ]
74. Kantaputra PN, Kaewgahya M, Khemaleelakul U, et al.Enamel-renal-gingival syndrome and FAM20A mutations. Am J Med Genet A 2014;164A(1):1-9. [ Links ]
75. Aspinall A, Raj S, Jugessur A, et al.Expanding the cleft phenotype: the dental characteristics of unaffected parents of Australian children with non-syndromic cleft lip and palate. Int J Paediatr Dent 2014;24(4):286-92. [ Links ]
76. Martins RB, de Souza RS, Giovani EM. Cleidocranial dysplasia: report of six clinical cases. Spec Care Dentist 2014;34(3):144-50. [ Links ]
77. Chalakkal P, Krishnan R, De Souza N, Da Costa G. A rare occurrence of supplementary maxillary lateral incisors and a detailed review on supernumerary teeth. J Oral Maxillofac Pathol 2018;22(1):149. [ Links ]

