










Services on Demand
Journal

Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkArchivos argentinos de pediatría
Print version ISSN 0325-0075On-line version ISSN 1668-3501
Arch. argent. pediatr. vol.117 no.3 Buenos Aires June 2019
http://dx.doi.org/10.5546/aap.2019.e274
CASE REPORT
http://dx.doi.org/10.5546/aap.2019.eng.e274
FKBP14 kyphoscoliotic Ehlers Danlos syndrome in adolescent patient: the first Colombian report
Felipe Ruiz-Boteroa, MD; Diana Ramirez-Montanoa, MD and Harry Pachajoa a,b, MD, PhD.
a. Universidad Icesi, Facultad de Ciencias de la Salud, Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER). Cali, Colombia. b. Departamento de Pediatría, Fundación Valle del Lili, Cali, Colombia.
E-mail address: Felipe Ruiz Botero, M.D.: Fruiz@icesi.edu.co
Funding: None.
Conflict of interest: None.
Received: 7-18-2018
Accepted: 12-6-2018
ABSTRACT
Ehlers-Danlos syndrome (EDS) is a group of clinically and genetically heterogeneous inherited connective tissue disorders, characterized by skin hyperextensibility, poor wound healing, joint hypermobility and tissue friability. Since 1997 a new spectrum of novel rare EDS-variants has been described, among which is included the EDS kyphoscoliotic type, characterized by severe muscular hypotonia at birth, severe progressive kyphoscoliosis, osteopenia, fragile eyeballs and vascular fragility. This EDS variant is caused by mutations in the PLOD1 gene; however, a rare recessive variant that compromises the FKBP14 gene has been reported, with additional clinical findings that includes gross motor developmental delay, myopathy, hearing impairment and a normal ratio of lysyl pyridinoline to hydroxylysyl pyridinoline in urine. We report the first Colombian patient with a FKBP14 c.362dupC mutation, with clinical features that include generalized hypotonia, delayed gross motor milestones, hearing loss, early-onset progressive kyphoscoliosis, joint hypermobility and foot deformities.
Key words: Ehlers Danlos syndrome; Spinal curvature; Molecular pathology.
INTRODUCTION
Ehlers-Danlos syndrome (EDS) is a group of clinically and genetically heterogeneous inherited connective tissue disorders, characterized by skin hyperextensibility, poor wound healing, joint hypermobility and tissue friability.1-3 Six EDS subtypes are recognized in the Villefranche nosology;1 however, since 1997 a new spectrum of novel rare EDS-variants have been described.2,4
Among these subtypes of EDS is the kyphoscoliotic type (MIM #225400), previously known as EDS VIA, which is characterized by severe muscular hypotonia at birth, joint hypermobility, luxations, severe progressive kyphoscoliosis, severe skin hyperelasticity, marfanoid habitus, osteopenia, fragile eyeballs associated with ocular globe rupture and occasionally vascular fragility.1 This EDS variant is caused by deficiencies of collagen-modifying enzyme procollagen-lysine 2-oxoglutarate 5-dioxygenase 1 (PLOD1), produced by a homozygous or compound heterozygous mutation of PLOD1 alleles.5
In 2012, six affected individuals with a rare recessive variant of kyphoescoliotic EDS whose clinical characteristics additionally included gross motor developmental delays; myopathy (confirmed by muscle MRI, histology and electron microscopy); hearing impairment (predominantly sensorineural), and a normal ratio of lysyl pyridinoline to hydroxylysyl pyridinoline in urine were reported.6 A homozygous frameshift mutation was identified as the causative factor. This mutation produces a translational frameshift and a premature stop codon in FKBP14.6 This subtype of EDS is currently called FKBP14-kyphoscoliotic EDS (MIM #614557) by the 2017 International Nosology of EDS.6,7
The present work reports the first case of a Colombian patient with molecular diagnosis of FKBP14-kyphoscoliotic EDS, utilizing next generation sequencing techniques which revealed a previously reported homozygous mutation in FKBP14 gene: c.362dupC (p.glu122Argfs*7).
CASE REPORT
We report the case of a 17-year-old girl of mixed race ancestry, originally from the southwest of Colombia. The patient was the product of the first pregnancy of consanguineous first grade cousins. Both parents reported no significant past medical history. During pregnancy, her mother experienced severe preeclampsia, and underwent a caesarean section at 34 weeks of gestation. At birth, the patient's weight was of 1900 gr (29th percentile) and 42 cm of height (23rd percentile). During the newborn physical assessment, generalized hypotonia, congenital bilateral hip dislocation and a neonatal gastric perforation were noted; which required management in the intensive care unit for two months. Orotracheal intubation was required during the first month of life.
Further assessment during this period revealed feeding difficulties, hiatal hernia and symptoms of gastroesophageal reflux (findings associated with her underlying pathology). Later, during early childhood, the patient exhibited delayed psychomotor development, sitting at 2 years of age, crawling at 3 years and ambulating at 5 years. Due to hip and ankle instability she required the use of leg braces until 5 years of age. Progressive kyphoescoliosis was noticed at 2 years of age, requiring surgical interventions at ages 10 and 15. Other findings included pectum excavatum and ankle luxation which were treated at age 15 (see Figure 1) and moderate left-sided sensorineural hearing loss, diagnosed at age 16.
Figure 1. 3D computed tomography reconstruction of thoracolumbar vertebrae
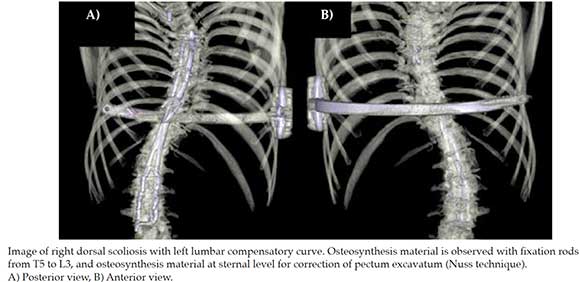
Our patient underwent initial outpatient genetic consultation at 15 years of age. During the physical assessment it was evidenced: weight of 39.2 kg (<1st percentile), height of 1.36 m (<1st percentile), generalized muscle hypotonia, myopathic gait, mid-facial hypoplasia, light blue sclera, long philtrum, micrognathia and progressive severe scoliosis (see Figure 2). Additional findings included severe shoulder, finger, wrist, ankle and toe joint hypermobility according to Beighton score (9/9);8 pectus excavatum, general xerosis, wrinkled skin on her palms and soles (see Figure 3), easy bruising, multiple isolated comedones and hypertrophic scaring, particularly on her back due to previous surgical interventions (see Figure 4).
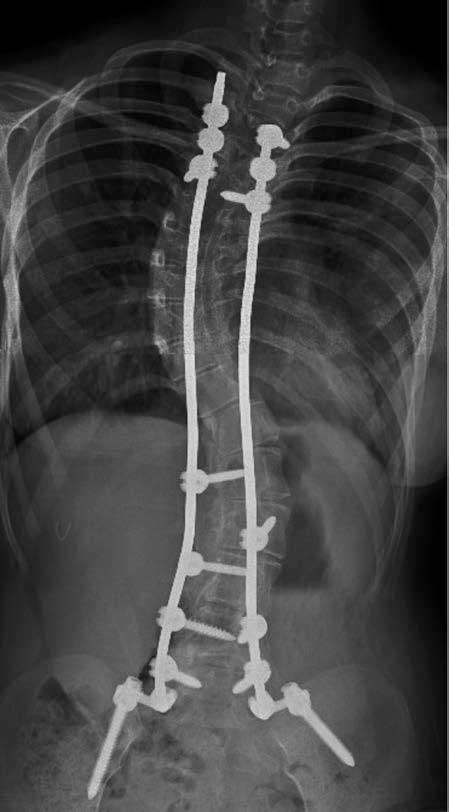
Figure 2. Anteroposterior full-length spinal X-Ray imaging
Image taken after surgical correction of scoliosis. Notice
the cervico-dorsal scoliotic curve with left convexity. Postsurgical
changes with rod-type osteosynthesis material and
transpedicular screws in the vertebral bodies T2, T3 and T4
in the upper segment; and L1 to L5 and sacral alierons in the
lower segment.
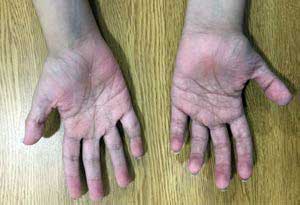
Figure 3. Palms of the hands with wrinkled skin and criss-cross
dermatoglyphic patterns
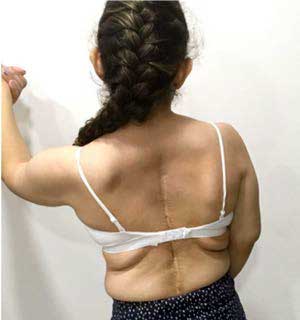
Figure 4. Back of the patient with multiple isolated
comedones and hypertrophic scaring due to previous
surgical interventions
Complementary studies included transthoracic echocardiogram, visual screening, cerebral magnetic resonance angiography and cerebral and paranasal computed tomography, all of which were within normal parameters. Spirometry revealed a restrictive lung disease pattern. Next-generation sequencing panel for Ehlers-Danlos syndrome reported a homozygous pathogenic variation of the FKBP14 gene, presenting an insertion of one cytosine residue within a 5C-nucleotide repeat in exon 3 (c.362dup). This produced a translational frameshift and a premature stop codon.
Parents received genetic counseling, while preventive follow-up for complications associated with the EDS syndrome was decided to be performed with the patient, among which are included routine assessment for inguinal hernia detection, ophthalmological follow-up for management and early diagnosis of glaucoma and/or retinal detachment and monitoring by neumology service due to the presence of pulmonary restrictive pattern in a patient surgically treated for kyphoscoliosis. No echocardiographic follow-up was performed during the last control, due to previous study within normal parameters; additionally, the patient continues observation by the orthopaedic surgery service.
DISCUSSION
We report the first case of a Colombian patient with kyphoescoliotic EDS. This subtype of EDS has been generally associated with homozygous mutations in the PLOD1 gene, who encodes the lysyl hydroxylase enzyme; however, since its first description in 2012,6 some cases with homozygous or compound heterozygous mutation in FKBP14 have been reported in the literature for this form of Ehlers Danlos.9-13
Aldeeri et al. in 2014, reported the case of a 3 year-old boy with a milder phenotype and a novel homozygous splice-site mutation in FKBP14. Due to the redundant umbilical skin found in this patient, they propose this sign as a potential early clinical feature for the diagnosis of this condition.9 Murray et. al, during the same year, reported the case of a 42 year-old American man born to nonconsanguineous parents, with a phenotype that included severe kyphoscoliosis, restrictive lung disease, short stature, mild hearing loss, decreased muscle mass and vascular findings that included a dissection of the celiac artery at age 41 and the complete occlusion of the superior mesenteric artery with compensatory flow through an enlarged and tortuous inferior mesenteric artery.10
Dordoni et al., in 2016, described the case of an 8 year-old Italian male with a compound heterozygous for a recurrent and a novel FKBP14 mutation, with a phenotype similar to that described in previous patients. This patient exhibited a pseudoaneurysm in the hypogastric artery that ruptured at 6 years of age and was safely treated with an endovascular procedure.11 In 2017, Bursztejn et. al characterized the cutaneous phenotype of an adolescent female patient with features that included multiple isolated comedones and molluscoid pseudotumours. Pseudotumours have been described as a distinctive finding in patients with classic EDS, but are rarely reported in other variants.12 Lately, Giunta et al., reported on 17 additional individuals from 15 unrelated families diagnosed with kyphoescoliotic EDS, representing the largest cohort of FKBP14-kyphoscoliotic EDS.13
Recently, the criteria for kyphoescoliotic EDS caused by pathogenic variants in either PLOD1 or FKBP14 have been revised. Presently, the major criteria includes (I) congenital muscle hypotonia, (II) congenital or early onset kyphoscoliosis (progressive or non-progressive) and (III) generalized joint hypermobility with dislocations/subluxations. Skin hyperextensibility and rupture/aneurysm of a medium size artery are included in the minor criteria.7 D'hondt et al., reviewed the vascular complications of vascular phenotypes, in nonvascular subtypes of EDS, and found that arterial dissections are most frequently reported in FKBP14-kyphoscoliotic EDS.14
In the present report, our patient fulfilled all three major criteria for kyphoescoliotic EDS. At present, no vascular complications have been detected; however, due to the different reports found in the literature, we believe that screenings for vascular anomalies, in addition to dilatation of the aortic root, are justified. Despite this condition being classified as a non-vascular type of EDS, the risk of arterial dissections and aneurysms supports a vascular surveillance approach until a better-characterized FKBP14-kyphoscoliotic EDS vascular phenotype indicates otherwise.
The FKBP14 c.362dupC mutation was previously reported in the literature in patients of Italian, French, English, German and Turkish descent.6, 10 This report contributes to the possibility that the recurrent c.362dupC mutation implicates small insertions or deletions in homopolymer regions, due DNA slippage, as previously discussed by Murray et al.10 Further research and detailed phenotypical and molecular description of new unreported cases, will allow for a better understanding of the pathogenesis and underlying mechanisms by which mutations in this gene occur.
1. Beighton P, De Paepe A, Steinmann B, TsipourasP, et al. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am J Med Genet. 1997; 77(1):31-7. [ Links ]
2. Bowen JM, Sobey GJ, Burrows NP, Colombi M, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017; 175(l):27-39. [ Links ]
3. Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, et al. The Ehlers-Danlos syndromes, rare types. Am J Med Genet C Semin Med Genet. 2017; 175(1):70-115. [ Links ]
4. Colombi M, Dordoni C, Chiarelli N, Ritelli M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am JMed Genet C Semin Med Genet. 2015; 169C(1):6-22. [ Links ]
5. Rohrbach M, Vandersteen A, Yis U, Serdaroglu G, et al. Pheno-typic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): Clinical, molecular, and biochemical delineation. Orphanet J Rare Dis. 2011; 6:46. [ Links ]
6. Baumann M, Giunta C, Krabichler B, Rüschendorf F, et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am J Hum Genet. 2012; 90(2):201-16. [ Links ]
7. Malfait F, Francomano C, Byers P, Belmont J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017; 175(1):8-26. [ Links ]
8. Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973; 32(5):413-8. [ Links ]
9. Aldeeri AA, Alazami AM, Hijazi H, Alzahrani F, et al. Excessively redundant umbilical skin as a potential early clinical feature of Morquio Syndrome and FKBP14-related Ehlers-Danlos syndrome. Clin Genet. 2014; 86(5):469-72. [ Links ]
10. Murray ML, Yang M, Fauth C, Byers PH. FKBP14-related Ehlers-Danlos syndrome: expansion of the phenotype to include vascular complications. Am J Hum Genet A. 2014; 164A(7):1750-5. [ Links ]
11. Dordoni C, Ciaccio C, Venturini M, Calzavara-Pinton P, et al. Further delineation of FKBP14-related Ehlers-Danlos syndrome: A patient with early vascular complications and non-progressive kyphoscoliosis, and literature review. Am J Med Genet A. 2016; 170(8):2031-8. [ Links ]
12. Bursztejn AC, Baumann M, Lipsker D. Ehlers-Danlos syndrome related to FKBP14 mutations: detailed cutaneous phenotype. Clin Exp Dermatol. 2017; 42(1):64-7. [ Links ]
13. Giunta C, Baumann M, Fauth C, Lindert U, et al. A cohort of 17 patients with kyphoscoliotic Ehlers-Danlos syndrome caused by biallelic mutations in FKBP14: expansion of the clinical and mutational spectrum and description of the natural history. Genet Med. 2017; 20(1):42-54. [ Links ]
14. D'hondt S, Van Damme T, Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: a systematic review. Genet Med. 2018; 20(6):562-73. [ Links ]

