










Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArchivos argentinos de pediatría
versão impressa ISSN 0325-0075versão On-line ISSN 1668-3501
Arch. argent. pediatr. vol.117 no.3 Buenos Aires jun. 2019
http://dx.doi.org/10.5546/aap.2019.e274
PRESENTACIÓN DE CASOS CLÍNICOS
http://dx.doi.org/10.5546/aap.2019.e274
Sindrome de Ehlers-Danlos dfoescoliotico-FKBP14 en una paciente adolescente: primer reporte de caso colombiano
Dr. Felipe Ruiz-Boteroa, Dra. Diana Ramírez-Montañoa y Dr. Harry Pachajoaa,b
a. Universidad Icesi, Facultad de Ciencias de la Salud, Centro de Investigaciones en Anomalías Congénitas y Enfermedades Raras (CIACER).
b. Departamento de Pediatría, Fundación Valle del Lili, Cali, Colombia.
Correspondencia: Dr. Felipe Ruiz Botero: Fruiz@icesi.edu.co
Financiamiento: Ninguno.
Conflicto de intereses: Ninguno que declarar.
Recibido: 18-7-2018
Aceptado: 6-12-2018
RESUMEN
El síndrome de Ehlers-Danlos es un conjunto de trastornos hereditarios del tejido conectivo, clínica y genéticamente heterogéneos, caracterizados por hiperextensibilidad cutánea, pobre cicatrización, hipermovilidad articular y friabilidad tisular. Desde 1997, se han reportado variantes poco frecuentes del síndrome, entre las cuales se incluye el de tipo cifoescoliótico, causado por mutaciones en el gen PLOD1, caracterizado por hipotonía muscular grave al nacer, cifoescoliosis grave progresiva, osteopenia, ojos frágiles y fragilidad vascular. También ha sido descrita una rara variante recesiva que compromete el gen FKBP14, con hallazgos clínicos adicionales, que incluyen retardo del desarrollo psicomotor, miopatía, hipoacusia y una proporción normal de lisil-piridinolina a hidroxilisil-piridinolina en la orina. Se presenta el primer caso de una paciente colombiana con una mutación FKBP14 c.362dupC, caracterizada por hipotonía generalizada, retardo en el desarrollo de los hitos motores gruesos, hipoacusia, cifoescoliosis progresiva temprana, hipermovilidad articular y deformidades en los pies.
Palabras clave: Síndrome de Ehlers-Danlos; Curvaturas de la columna vertebral; Patología molecular.
INTRODUCCIÓN
El síndrome de Ehlers-Danlos (Ehlers-Danlos syndrome; EDS, por sus siglas en inglés) es un grupo de trastornos hereditarios del tejido conectivo, clínica y genéticamente heterogéneos, caracterizados por hiperextensibilidad cutánea, pobre cicatrización de heridas, hipermovilidad articular y friabilidad de los tejidos.1-3 Seis subtipos de EDS son reconocidos en la nosología de Villefranche;1 sin embargo, desde 1997, un nuevo espectro de variantes infrecuentes del EDS ha sido descrito.2,4
Entre estos subtipos de EDS, se incluye el tipo cifoescoliótico (MIM #225400), previamente conocido como EDS VIA, caracterizado por hipotonía muscular grave al nacer, hipermovilidad articular, luxaciones, cifoescoliosis grave progresiva, hiperelasticidad cutánea, hábito marfanoide, osteopenia, ojos frágiles asociados con la ruptura del globo ocular y, ocasionalmente, fragilidad vascular.1 Esta variante de EDS es causada por la deficiencia de procolágeno-lisina 2-oxa-glutarato 5-dioxigenasa 1 (PLOD1), producida por una mutación en homocigosis o heterocigosis compuesta de los alelos PLOD1.5
En 2012, se reportaron seis individuos afectados con una rara variante recesiva de EDS, cuyas características clínicas, adicionalmente, incluían retardo del desarrollo psicomotor, miopatía (confirmada por resonancia magnética nuclear -RMN-, histología y microscopía electrónica), hipoacusia (predominantemente, neurosensorial) y una proporción normal de lisil-piridinolina a hidroxilisil-piridinolina en la orina.6 Se identificó una mutación homocigota que producía un desplazamiento traslacional del marco de lectura y un codón de parada prematuro en el gen FKBP14.6 Este subtipo de EDS, actualmente, es nombrado EDS cifoescoliótico-FKBP14 (MIM #614557) por la nosología internacional de EDS de 2017.6,7
El presente trabajo reporta el primer caso de una paciente colombiana con diagnóstico molecular de EDS cifoescoliótico-FKBP14, en la que se utilizan técnicas de secuenciación de siguiente generación y se se evidencia una mutación homocigota en el gen FKBP14: c.362dupC (p.glu122Argfs*7), previamente descrita en la literatura.
REPORTE DE CASO
Se reporta el caso de una joven de 17 años de ascendencia mixta, originaria del suroeste colombiano. La paciente era producto de la primera gesta de primos consanguíneos en primer grado. Ambos padres informaron que no había antecedentes médicos significativos. Durante el embarazo, la madre presentó preeclampsia grave, por lo que se le realizó una cesárea a las 34 semanas de gestación. Al nacer, la paciente presentó un peso de 1900 g (percentil 29) y talla de 42 cm (percentil 23). Durante el examen físico del recién nacido, se evidenció hipotonía generalizada, luxación congénita de cadera bilateral y perforación gástrica neonatal (complicación conocida de la EDS), que requirió el manejo en la Unidad de Cuidados Intensivos durante dos meses con intubación orotraqueal durante el primer mes de vida.
Las evaluaciones adicionales durante este período revelaron dificultad para la alimentación, hernia hiatal y reflujo gastroesofágico (hallazgos asociados a su patología de base). Posteriormente, durante la infancia temprana, se identificó retardo en el desarrollo psicomotor y presentó la sedestación completa a los 2 años de edad, gateo a los 3 años y deambulación independiente a los 5 años. Debido a la inestabilidad de la cadera y del tobillo, la paciente requirió el uso de férulas ortopédicas en los miembros inferiores hasta los 5 años de edad. La cifoscoliosis progresiva fue percibida a los 2 años de edad y requirió una operación quirúrgica a los 10 y a los 15 años.
Otros hallazgos incluyeron pectum excavatum y luxación del tobillo, que fueron tratados quirúrgicamente a los 15 años e hipoacusia moderada izquierda neurosensorial, diagnosticada a los 16 años de edad.
La paciente asistió a la consulta externa de Genética Médica a los 15 años de edad. Durante la evaluación, se evidenció peso de 39,2 kg (< percentil 1), altura de 1,36 m (< percentil 1), hipotonía muscular generalizada, marcha miopática, hipoplasia del tercio medio facial, escleras azules, filtro largo, micrognatia y escoliosis grave progresiva (Figura 2). Adicionalmente, se identificó hipermovilidad articular grave de los hombros, de los dedos de la mano, de la muñeca, del tobillo y de los dedos de los pies, con una clasificación, según la Escala de Beighton, de 9/9,8 pectus excavatum, xerosis generalizada, piel arrugada en las plantas y en las palmas (Figura 3), aparición de hematomas con facilidad, presencia de múltiples comedones aislados y cicatrización hipertrófica, particularmente, en el tronco, debido a operaciones quirúrgicas previas (Figura 4).

Figura 1. Reconstrucción 3D de la tomografía axial computarizada de columna tóraco-lumbar
Imagen de escoliosis dorsal derecha con curva compensatoria lumbar izquierda. Se observa material de osteosíntesis con varillas de fijación de T5 a L3 y material de osteosíntesis a nivel esternal para corrección de pectum excavatum (Técnica de Nuss).
A. Vista posterior. B. Vista anterior.
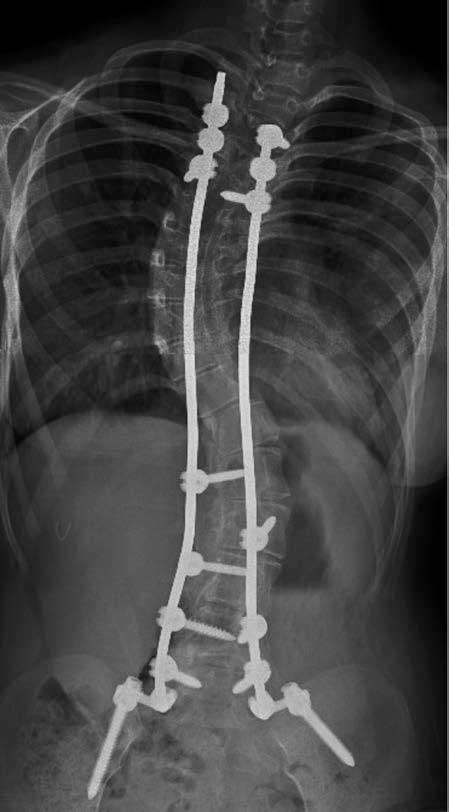
Figura 2. Radiografía de columna total anteroposterior
Imagen tomada después de la corrección quirúrgica de la escoliosis. Nótese la curva escoliótica cervicodorsal con convexidad izquierda. Cambios posquirúrgicos con material de osteogénesis tipo barra y tornillos transpediculares en los cuerpos vertebrales T2, T3 y T4 en el segmento superior y en el segmento inferior de L1 a L5 y alas del sacro
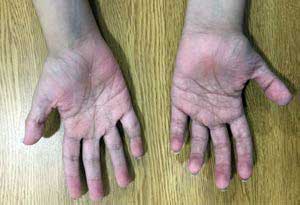
Figura 3. Nótese la piel arrugada y dermatoglifos en los cruces, en las palmas
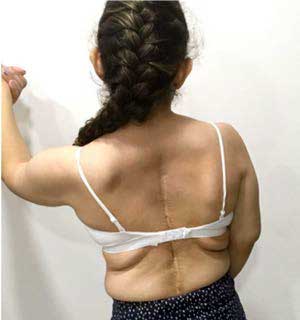
Figura 4. Dorso de la paciente con múltiples comedones aislados y cicatrización hipertrófica debido a operaciones quirúrgicas previas
Los estudios complementarios incluyeron ecocardiograma transtorácico, tamizaje visual, angiografía por resonancia magnética y tomografía axial computarizada cerebral y paranasal, con resultados dentro de los parámetros de normalidad. La espirometría reveló un patrón restrictivo de enfermedad pulmonar. Se realizó panel molecular de siguiente generación para el EDS, que evidenció una variante patogénica en homocigosis para el gen FKBP14, correspondiente a la inserción de un residuo de citosina dentro de una repetición de nucleótido 5C en el exón 3(c.362dup), con el consecuente cambio traslacional del marco de lectura y un codón prematuro de parada.
Los padres recibieron asesoramiento genético, mientras que, con la paciente, se decidió realizar un seguimiento preventivo de complicaciones asociadas al EDS, entre lo que se incluyó la evaluación rutinaria para la detección de hernia inguinal, la interconsulta con el Servicio de Oftalmología para el manejo y el diagnóstico temprano de glaucoma y/o desprendimiento de
retina, y el seguimiento por parte de Neumología del patrón restrictivo pulmonar en la paciente con operación quirúrgica de su cifoscoliosis. No se realizó una ecocardiografía durante el último control por presentar un estudio previo normal. Adicionalmente, la paciente continuaba el monitoreo por parte del Servicio de Cirugía Ortopédica.
DISCUSIÓN
Se presenta el caso de una paciente colombiana con EDS cifoescoliótico. Este subtipo ha sido, generalmente, asociado con mutaciones en homocigosis del gen PLOD1, que codifica la enzima lisil hidroxilasa; sin embargo, desde su primera descripción en 2012,6 algunos casos con mutaciones en homocigosis o heterocigotos compuestos en el gen FKBP14 han sido reportados en la literatura para esta forma de EDS.9-13
Aldeeri y col., en 2014, publicaron el caso de un niño de 3 años de edad con un fenotipo leve y una nueva mutación homocigota del sitio de splicing en FKBP14. Debido a la presencia de tejido redundante umbilical (ombligo cutáneo) en el paciente, ellos propusieron este signo como un potencial hallazgo clínico temprano para el diagnóstico de esta enfermedad.9 Murray y col., durante el mismo año, reportaron el caso de un hombre de 42 años de origen norteamericano, nacido de padres no consanguíneos, con un fenotipo que incluía cifoscoliosis grave, enfermedad pulmonar restrictiva, baja talla, hipoacusia leve, reducción de la masa muscular y hallazgos vasculares, como disección de la arteria celíaca a los 41 años y oclusión completa de la arteria mesentérica superior con flujo compensatorio mediante una mesentérica inferior agrandada y tortuosa.10
Dordoni y col., en 2016, describieron el caso de un niño italiano de 8 años de edad con una mutación heterocigota compuesta para una mutación recurrente y otra nueva en FKBP14, con un fenotipo similar al descrito en pacientes anteriores. Este presentaba un pseudoaneurisma en la arteria hipogástrica que se rompió a los 6 años de edad, que fue tratado mediante procedimiento endovascular.11 En 2017, Bursztein y col. caracterizaron el fenotipo cutáneo de una paciente adolescente con presencia de múltiples comedones aislados y pseudotumores moluscoides. Los pseudotumores han sido descritos como un hallazgo distintivo de los pacientes con EDS clásico, pero han sido, rara vez, reportados en otras variantes.12 Ultimamente, se publicaron 17 casos adicionales de 15 familias no relacionadas, con EDS cifoescoliótico, que representó la cohorte más grande de EDS cifoescoliótico-FKBP14.13
Recientemente, los criterios para EDS cifoescoliótico causados por variantes patogénicas en PLOD1 o FKBP14 han sido revisados. En la actualidad, los criterios mayores incluyen lo siguiente: (I) hipotonía congènita; (II) cifoescoliosis congénita o de aparición temprana (progresiva o no progresiva); (III) hipermovilidad articular generalizada con dislocaciones/subluxaciones. La hiperextensibilidad cutánea y la ruptura/aneurisma de una arteria de mediano calib re se incluyen dentro de los criterios menores.7 Una revisión de las complicaciones vasculares, en los tipos no vasculares de EDS, halló que las disecciones arteriales eran las más frecuentes en EDS cifoescoliótico-FKBP14.14
En el presente reporte, nuestra paciente cumplió los tres criterios principales para EDS cifoescoliótico. Hasta el momento, no se evidenciaron complicaciones vasculares; sin embargo, debido a varios reportes en la literatura, se cree que el tamizaje para anomalías vasculares, adicional a la dilatación de la raíz aórtica, se encuentra justificado. A pesar de que esta enfermedad se encuentra clasificada dentro de los tipos no vasculares de EDS, el riesgo de presentar aneurismas o disección arterial apoya un enfoque de vigilancia vascular hasta que una mejor caracterización del fenotipo vascular de EDS cifoescoliótico-FKBP14 indique lo contrario.
La mutación FKBP14 c.362dupC ha sido previamente reportada en la literatura, en pacientes de descendencia italiana, francesa, inglesa, alemana y turca.610 Este caso contribuye a la posibilidad de que la mutación recurrente c.362dupC involucre pequeñas inserciones o deleciones en regiones homopolímeras, debido al deslizamiento de ácido desoxirribonucleico (ADN) (slippage), como fue previamente descrito.10 Una investigación adicional y la detallada descripción fenotípica y molecular de nuevos casos permitirán una mejor comprensión de los mecanismos subyacentes y la patogénesis mediante la cual ocurren las mutaciones en este gen.
1. Beighton P, De Paepe A, Steinmann B, TsipourasP, et al. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Am J Med Genet. 1997; 77(1):31-7. [ Links ]
2. Bowen JM, Sobey GJ, Burrows NP, Colombi M, et al. Ehlers-Danlos syndrome, classical type. Am J Med Genet C Semin Med Genet. 2017; 175(l):27-39. [ Links ]
3. Brady AF, Demirdas S, Fournel-Gigleux S, Ghali N, et al. The Ehlers-Danlos syndromes, rare types. Am J Med Genet C Semin Med Genet. 2017; 175(1):70-115. [ Links ]
4. Colombi M, Dordoni C, Chiarelli N, Ritelli M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am JMed Genet C Semin Med Genet. 2015; 169C(1):6-22. [ Links ]
5. Rohrbach M, Vandersteen A, Yis U, Serdaroglu G, et al. Pheno-typic variability of the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VIA): Clinical, molecular, and biochemical delineation. Orphanet J Rare Dis. 2011; 6:46. [ Links ]
6. Baumann M, Giunta C, Krabichler B, Rüschendorf F, et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am J Hum Genet. 2012; 90(2):201-16. [ Links ]
7. Malfait F, Francomano C, Byers P, Belmont J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017; 175(1):8-26. [ Links ]
8. Beighton P, Solomon L, Soskolne CL. Articular mobility in an African population. Ann Rheum Dis. 1973; 32(5):413-8. [ Links ]
9. Aldeeri AA, Alazami AM, Hijazi H, Alzahrani F, et al. Excessively redundant umbilical skin as a potential early clinical feature of Morquio Syndrome and FKBP14-related Ehlers-Danlos syndrome. Clin Genet. 2014; 86(5):469-72. [ Links ]
10. Murray ML, Yang M, Fauth C, Byers PH. FKBP14-related Ehlers-Danlos syndrome: expansion of the phenotype to include vascular complications. Am J Hum Genet A. 2014; 164A(7):1750-5. [ Links ]
11. Dordoni C, Ciaccio C, Venturini M, Calzavara-Pinton P, et al. Further delineation of FKBP14-related Ehlers-Danlos syndrome: A patient with early vascular complications and non-progressive kyphoscoliosis, and literature review. Am J Med Genet A. 2016; 170(8):2031-8. [ Links ]
12. Bursztejn AC, Baumann M, Lipsker D. Ehlers-Danlos syndrome related to FKBP14 mutations: detailed cutaneous phenotype. Clin Exp Dermatol. 2017; 42(1):64-7. [ Links ]
13. Giunta C, Baumann M, Fauth C, Lindert U, et al. A cohort of 17 patients with kyphoscoliotic Ehlers-Danlos syndrome caused by biallelic mutations in FKBP14: expansion of the clinical and mutational spectrum and description of the natural history. Genet Med. 2017; 20(1):42-54. [ Links ]
14. D'hondt S, Van Damme T, Malfait F. Vascular phenotypes in nonvascular subtypes of the Ehlers-Danlos syndrome: a systematic review. Genet Med. 2018; 20(6):562-73. [ Links ]

