










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos argentinos de pediatría
versión impresa ISSN 0325-0075versión On-line ISSN 1668-3501
Arch. argent. pediatr. vol.117 no.4 Buenos Aires ago. 2019
http://dx.doi.org/10.5546/aap.2019.271
ARTÍCULO ESPECIAL
http://dx.doi.org/10.5546/aap.2019.271
Enfermedad de Pompe infantil: Diagnóstico y tratamiento
Dra. Luisa B. Baya, Dra. Inés Denzlerb, Dra. Consuelo Durandc, Dr. Hernán Eiroad, Bioquímico MSC Joaquín Frabasilc, Dr. Alejandro Fainboime, Dra. Clarisa Maxitb, Farmacéutica Andrea Schenonec y Dra. Norma Spécolaf
a. Consultora en el Hospital Garrahan.
b. Servicio de Neurología Infantil, Hospital Italiano de Buenos Aires.
c. Laboratorio Chamoles.
d. Servicio de Errores Congénitos del Metabolismo, Hospital Garrahan.
e. Hospital de Día Polivalente, Hospital de Niños Ricardo Gutiérrez.
f. Unidad de Metabolismo, Hospital Sor María Ludovica, La Plata. Buenos Aires.
Correspondencia: Dra. Luisa B. Bay: bay.luisa@gmail.com
Financiamiento: Ninguno.
Conflicto de intereses: La Dra. Luisa B. Bay y el Dr. Alejandro Fainboim han recibido honorarios por actividad docente del laboratorio que comercializa la medicación para la enfermedad que se actualiza.
Recibido: 10-7-2018
Aceptado: 27-12-2018
RESUMEN
La enfermedad de Pompe, o deficiencia de maltasa ácida o glucogenosis tipo II, es una grave enfermedad genética, autosómica recesiva, progresiva, poco frecuente, causada por la deficiencia en la enzima alfa glucosidasa.
En la edad pediátrica, puede presentarse con la "forma clásica", la más conocida, con grave compromiso cardíaco y franca hipotonía, o con la "forma no clásica", con comienzo temprano del compromiso motor. La "forma de comienzo tardío" del adulto también puede ocurrir en la infancia o en la adolescencia.
Se actualizan los hallazgos clínicos y de diagnóstico disponibles, ya que un tratamiento temprano con reemplazo de la enzima faltante puede mejorar la supervivencia y la calidad de vida del paciente. Se revisan los beneficios y los efectos adversos del tratamiento disponible y nuevas líneas de investigación terapéutica.
Palabras clave:: Enfermedad de Pompe; Glucogenosis tipo II; Miocardiopatías; Hipotonía muscular; Trastornos motores.
INTRODUCCIÓN
La enfermedad de Pompe (EP), conocida también como deficiencia de maltasa ácida o glucogenosis tipo II, es una afección genética, autosómica recesiva, grave, progresiva, poco frecuente, causada por la deficiencia en la enzima alfa glucosidasa ácida (AGA).1
Reconocida como el primer desorden de almacenamiento lisosomal, fue descrita por Johannes Pompe, patólogo holandés, en 1932, en una niña fallecida con grave debilidad muscular y miocardiopatía hipertrófica, con depósito de glucógeno en el corazón, en el hígado, en los riñones y en el músculo esquelético.2 En 1979, Hers3 demostró que la acumulación de glucógeno la producía la deficiencia de maltasa ácida en los lisosomas.
Se identificó el gen (GAA) de esta enzima en el brazo largo del cromosoma 17 y se han descrito más de 450 mutaciones.4
Tiene un amplio espectro de fenotipos, síntomas, edad de inicio, progresión y afectación de los distintos órganos. Se diferencian la EP infantil (EPI) y la de inicio tardío (EPT), y hay formas intermedias.
La incidencia combinada de todas las formas clínicas se estima de 1:40 000 a 1:300 000, dependiendo de la etnia y de las zonas geográficas estudiadas.5
DESCRIPCIÓN CLÍNICA Y EVOLUCIÓN NATURAL
Se adopta la denominación del American College of Medical Genetics (ACMG),6 que incluye la forma "infantil clásica" (EPIC), descrita con más frecuencia, con cardiomegalia grave, hepatomegalia, hipotonía desde el nacimiento y muerte temprana; la forma "infantil no clásica" (EPINC), que se manifiesta desde el primer año, pero con progresión más lenta y miocardiopatía menos grave;7,8 y la EPT, que se manifiesta en la niñez o en la juventud y, en el adulto, entre la segunda y la sexta década de la vida, con miopatía lentamente progresiva, por lo general, sin miocardiopatía.
La EPIC se presenta a la edad mediana de 2 meses (0-12) con dificultades alimentarias y falla de crecimiento en el 44-97 % de los casos, miocardiopatía en el 50-92 %, hipotonía en el 20-63 % y dificultad respiratoria en el 27-78 %.
La edad media de fallecimiento comunicada fue a los 8,7 meses (0,3-73), y la sobrevida a los 12 meses de edad fue del 25,7 %, y, para el grupo que sobrevivió sin asistencia respiratoria, del 16,9 %. A los 18 meses, la sobrevida fue del 12,3 % y del 6,7 %, respectivamente.7,9
Los primeros síntomas de la EPINC son el retraso en la maduración motora y la debilidad muscular con una edad mediana de presentación a los 2,6 años (0,5-13) y de diagnóstico a los 4 años (0-16).10
Manifestaciones neuromusculares
Se presentan entre los 0 y los 4 meses en la EPIC y son hipotonía desde el nacimiento con hipo- o arreflexia y debilidad en los músculos de la cara (facies característica), del tronco y de las extremidades. La mayor afectación es de los músculos proximales de las extremidades inferiores. Aparecen patrones de movimiento compensatorios por la gran debilidad y la escasa movilidad que causan deformidades y retracciones articulares. La debilidad de la musculatura orolingual y diafragmática afecta precozmente las funciones deglutoria y respiratoria, con retraso en el lenguaje expresivo.9-11
En la EPINC, son más afectados los músculos flexores del cuello, lo que dificulta levantar la cabeza desde la posición supina. La debilidad proximal en los miembros causa caídas frecuentes, dificultad para subir escaleras, para correr y practicar deportes. En el 50 % de los niños, se describe facies miopática, escoliosis, ausencia de reflejos. No se encontraron disfagia ni trastornos del lenguaje, salvo 1 caso.12
En la EPT (niños, jóvenes, adultos), hay disfunción muscular progresiva, de los músculos proximales de los miembros inferiores y paratroncales, luego del diafragma y de los músculos respiratorios accesorios.13
Manifestaciones respiratorias
En la EPIC, la insuficiencia respiratoria causa morbimortalidad temprana. La debilidad de los músculos respiratorios, la alteración del mecanismo de la tos y de la deglución provocan infecciones respiratorias de las vías altas y neumonías repetidas.14
En la EPINC, el 48 % de los niños tiene disminución de la capacidad vital forzada y, a los 9 años de edad mediana (rango: 5-16 años), requiere ventilación asistida o fallece.6
El trastorno respiratorio del sueño se manifiesta con letargia, irritabilidad, ronquidos nocturnos o apneas.15
Manifestaciones cardíacas
En la EPIC, la hipertrofia ventricular, predominante en la pared posterior del ventrículo izquierdo y septum interventricular, puede manifestarse aun intraútero. Puede haber obstrucción a la salida del ventrículo izquierdo. La evolución rápida y progresiva deriva en una miocardiopatía dilatada con insuficiencia cardíaca terminal. La fracción de eyección cae después de los 5 meses de edad. El depósito de glucógeno puede provocar trastornos de conducción, arritmias ventriculares graves, perfusión coronaria inadecuada, isquemia subendocárdica y riesgo de muerte súbita.16
El compromiso cardíaco en la EPINC se encontró solo en el 10 % de los casos.11
Manifestaciones gastrointestinales y nutricionales
En los lactantes, la hipotonía facial, la macroglosia y la debilidad motora oral causan dificultad para succionar, lo que afecta el crecimiento y la ganancia de peso. La debilidad muscular puede provocar disfagia, reflujo gastroesofágico, gastroparesia y estreñimiento. Puede haber hepatomegalia y/o esplenomegalia.14
Exámenes complementarios y métodos de diagnóstico (Tabla 1)
Tabla 1. Exámenes complementarios y métodos de diagnósticos

El daño muscular produce elevación conjunta de la creatina-fosfocinasa (CPK) (1500 UI o 2000 UI) y de las transaminasas hepáticas.17
• Electrocardiograma: revela la miocardiopatía con hipertrofia ventricular y trastornos del ritmo. Acortamiento PR, QRS muy amplio.618
• Electromiograma: en la EPI, muestra irritabilidad de membrana y actividad denervatoria, con descargas repetitivas de alta frecuencia. Puede haber descargas miotónicas, en ausencia de miotonía clínica. La contracción voluntaria muestra potenciales de unidad motora polifásicos breves y de baja amplitud.19 En la EPT, hay hallazgos compatibles con miopatía en los músculos proximales y/o actividad denervatoria en los paravertebrales.20
• Imágenes: la radiografía de tórax en la EPIC muestra cardiomegalia, reducción del volumen pulmonar y/o áreas de atelectasia. En la EPT, suele ser normal.7
La resonancia magnética (RM) es muy útil en la EPT; permite determinar el patrón y la extensión del compromiso muscular.21
Actividad enzimática: fundamental para el diagnóstico; se mide por fluorometría, colorimetría o por espectrometría de masa en tándem. Por su accesibilidad y rapidez, el primer test diagnóstico es en gotas de sangre en papel de filtro (GSPF). Un resultado positivo debe confirmarse en linfocitos, leucocitos, músculo o fibroblastos; este último es el gold standard del diagnóstico, pero es invasivo y el resultado puede demorar hasta 6 semanas.22 La metodología de la espectrometría de masa sugiere que la medición en sangre es comparable con la de fibroblastos.23-25
Estudios bioquímicos complementarios a la actividad enzimática
El tetrasacárido de glucosa (Glc4) es un biomarcador urinario complementario de la actividad enzimática y permite evaluar la respuesta al tratamiento. Se han reportado niveles francamente elevados de Glc4 en la EPI a diferencia de la EPT.26
Biopsia muscular
Una toma del sector más comprometido clínicamente podrá aportar a la hipótesis diagnóstica de EP u orientar un diagnóstico alternativo.27,28
En la EPIC, se presenta una grave miopatía vacuolar con acumulación de glucógeno, técnica de ácido peryódico de Schiff (periodic acid Schiff; PAS, por sus siglas en inglés) positiva y fosfatasa ácida positiva (marcador lisosomal y de autofagia). Existe disminución de miofibrillas, observable con miosina ATPasa. A nivel ultraestructural, se suele observar glucógeno intra- y extralisosomal, distorsión de la estructura con disminución notable de miofibrillas y vacuolas autofágicas.
En la EPT, puede haber biopsias normales o de características graves similares a las de la EPI.
Con una biopsia sugestiva de EP, debe confirmarse el diagnóstico con dosaje enzimático o de ácido desoxirribonucleico (ADN).29 Una biopsia normal o inespecífica no excluye el diagnóstico de EP.
Estudios moleculares (ADN)
El gen GAA, en el cromosoma 17q25, consta de 20 exones. Origina una proteína de 952 aminoácidos. De las más de 500 variantes de secuencia descritas, más de 350 presentan algún grado de patogenicidad, unas 90 fueron clasificadas como mutaciones no patogénicas y más de 90 tenían significado incierto.30 Las alteraciones incluyen mutaciones sin sentido, con cambio de sentido, que afectan el splicing,deleciones/inserciones, entre otras.4
Casi el 50 % de los pacientes caucásicos con EPT tienen la mutación intrónica c.-32-13T>G, que afecta el splicing, y nunca fue hallada en los pacientes con EPI, por lo que su presencia en uno de los alelos la excluiría.
No existe una estricta correlación genotipo-fenotipo, pero la presencia de mutaciones sin sentido en los dos alelos del gen, que producen una proteína trunca, se asocia con la EPIC.
Otros factores (genéticos y ambientales) pueden influir en el fenotipo, por lo que pacientes con las mismas mutaciones pueden presentar fenotipos clínicamente diferentes. El análisis genético, si el caso índice es conocido, permite el diagnóstico prenatal y la identificación de individuos portadores.31
Existe pseudodeficiencia de AGA, con actividad enzimática descendida in vitro, pero la actividad in vivo es suficiente para no desarrollar la enfermedad. El diagnóstico se hace por la presencia de las mutaciones c.1726G>A (p. G576S) y c.2065G>A (p. E689K) en el mismo alelo (en cis) y ambas en homocigosis.
Se aconseja estudiar a los familiares de los pacientes con diagnóstico confirmado, con riesgo, en especial, en los hermanos por su carácter recesivo y por la posibilidad de que la EP se mantenga silente por tiempo prolongado.32
Diagnósticos diferenciales (Tablas 2 y 3)
En la EPIC, la cardiomegalia y la hipotonía son los signos principales. La primera debe diferenciarse de miocarditis virales, miocardiopatía hipertrófica, enfermedades mitocondriales, de defectos de transporte de carnitina y de oxidación de ácidos grasos.
Tabla 2. Diagnósticos diferenciales en la forma "infantil clásica"

Tabla 3. Diagnósticos diferenciales en la forma infantil "no clásica" de inicio a partir del ano de edad
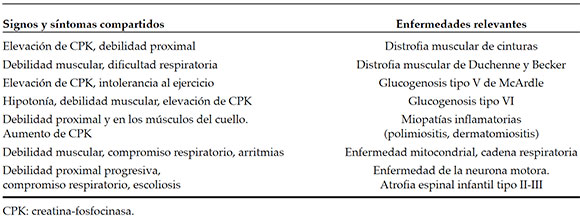
La hipotonía grave en un lactante debe diferenciarse de atrofia espinal infantil, distrofias musculares y miopatías congénitas. La EPT debe diferenciarse de otros desórdenes neuromusculares. Estudios complementarios ayudarán en el diagnóstico.
TRATAMIENTO Medidas generales
Si bien existe un tratamiento específico desde 2006, las medidas generales y paliativas siguen siendo esenciales:
• Vacunación completa, que incluye antineumocóccica y antigripal y profilaxis del virus sincicial respiratorio. Tratamiento intensivo y precoz de las afecciones respiratorias. Se debe estimular la succión y la fonación. Se debe mantener la alimentación oral o por sonda o gastrostomía, acorde a las necesidades del paciente.33
• Equipamiento auditivo en presencia de pérdida neurosensorial, que puede ocurrir en la EPI.34 Seguimiento por un cardiólogo con experiencia, ya que las drogas habituales pueden estar contraindicadas en alguna etapa de la enfermedad.6
• Se deben evitar posturas en flexión, mantener la alineación raquídea, ejercicios físicos, equipamiento ortésico o productos de apoyo si son necesarios.35,36
Tratamiento específico
El tratamiento específico disponible para la EP en todas las edades es el tratamiento de reemplazo enzimático (TRE) con alfa glucosidasa ácida recombinante humana (AGArh), alglucosidasa alfa (Myozyme®, Genzyme Corporation), que fue aprobado por la Food and Drug Administration (FDA) en Estados Unidos de América, en 2006,37,38 con un estudio con grupo control de 8 enfermos y por 52 semanas, que se complementó con los múltiples casos tratados publicados en los últimos 12 años10,16,31,39,40-43
Comparando con la evolución natural, en la EPIC con TRE precoz, mejoran la expectativa de vida y la función cardíaca, y disminuye la cardiomegalia.44 La mejoría del tono y de la fuerza muscular dependen de la precocidad del tratamiento y de la gravedad de la mutación. No siempre se adquiere una significativa independencia motora. La función respiratoria, la deglución y el crecimiento se estabilizan. La calidad de vida mejora, pero permanece afectada.45
Eficiencia del tratamiento.
Estado cross-reactive immunologic material
Antes de iniciar el TRE, se debe conocer si el paciente produce material inmunológicamente reactivo (cross-reactive inmunología material; CRIM, por sus siglas en inglés) o no, porque define la eficiencia del tratamiento.
Los pacientes con mutaciones nulas, de un 25 % a un 30 % de los casos de EPIC, no producen nada de enzima AGA, por lo que carecen de dicho material (CRIM negativo). En ellos, la enzima infundida será reconocida como una proteína extraña y producirá altos títulos de anticuerpos neutralizantes, que anularán su efecto. En estos casos con TRE, a la mejoría inicial puede seguirle un deterioro clínico significativo.46,47
Los pacientes que presentan algo de la proteína AGA propia, aunque inactiva, son CRIM positivos; la proteína AGArh infundida no se reconocerá como extraña y el título de anticuerpos se mantendrá bajo, con mayor eficiencia del tratamiento.
El estado CRIM se establece antes de empezar el TRE en sangre conservada a -80 °C o cultivo de fibroblastos. Ante dificultades en su determinación, será necesario el estudio de las mutaciones para inferir el estado CRIM.48
Inducción de la tolerancia inmune
En pacientes CRIM negativos, se puede promover la tolerancia inmune al TRE, idealmente, antes del comienzo del tratamiento en un centro capacitado, mediante el uso de drogas, como rituximab y metotrexato, con un soporte mensual de gammaglobulina. Esto deprime la respuesta inmune.49
Indicación del tratamiento de reemplazo enzimático
Se indica el TRE a todo paciente con EPI sintomática, con diagnóstico clínico y enzimático, con el estudio CRIM o el estudio genético relizado.38 La dosis es de 20 mg/kg cada dos semanas según las indicaciones de preparación e infusión de la compañía productora.29
No hay consenso amplio de que la dosis o la frecuencia mayores mejoren la supervivencia o la calidad de vida y sí podrían incrementarse los efectos adversos.50
No se indica en presencia de otras enfermedades potencialmente fatales ni en casos muy graves con ventilación invasiva por insuficiencia respiratoria no aguda.38
Es medicación de alto costo y, hasta tanto las autoridades de salud del país se expidan sobre su uso, se sugiere que solo se indique con el diagnóstico confirmado y con discusión con un grupo de profesionales con experiencia en la enfermedad y su tratamiento.
Efectos adversos del tratamiento de reemplazo enzimático
Como cualquier infusión de una proteína, puede provocar reacciones alérgicas, por anticuerpos inmunoglobulina G (IgG).51 Generalmente, son reacciones leves o moderadas y se controlan con la reducción de la velocidad de infusión o su interrupción temporal, y se restaura cuando las manifestaciones se resuelven, o se recurre a antihistamínicos y corticoides que pueden utilizarse como premedicación. Aisladamente, puede presentarse reacción anafiláctica grave, por anticuerpos inmunoglobulina E (IgE), lo que refuerza la necesidad de infusión en un área hospitalaria.
La familia del paciente deberá estar informada del beneficio real esperado y del riesgo de complicaciones.
Nuevos tratamientos
Otros tratamientos específicos52,53 están en desarrollo, como la terapia génica,54 el trasplante autólogo de células hematopoyéticas asociado a lentivirus,55 el uso de chaperonas,56 la TRE recombinante de segunda generación, la terapia de reducción de sustrato, pero no tienen aún demostración de beneficios clínicos.52,54
Nuevo fenotipo en pacientes con enfermedad de Pompe infantil clásica tratados con reemplazo enzimático
El TRE con AGA dio lugar a un nuevo fenotipo.35 No se trata de un cambio del fenotipo clásico al de inicio tardío, sino que presenta características clínicas particulares.
El mayor estudio en este sentido57 describe a 11 pacientes CRIM positivos de 5,4-12 años de edad, sintomáticos antes de los 6 meses, con actividad enzimática < al 1 %, sin requerimiento de soporte ventilatorio, que comenzaron el TRE antes de los 6 meses y cuya sobrevida fue mayor de 5 años. A los 5 meses del inicio del TRE, resolvieron su miocardiopatía. Cinco de los 11 pacientes presentaron algún tipo de arritmia cardíaca tratable. Ningún paciente necesitó soporte ventilatorio; dos de ellos tuvieron apneas obstructivas. Siete de los 11 pacientes alcanzaron la marcha independiente, y el resto, con algún tipo de soporte. La mayoría presentó debilidad residual generalizada asociada a hipotonía, predominante en los músculos del cuello, los proximales de los miembros inferiores (extensores de cadera, aductores y abductores), tibial anterior con dificultad en la dorsiflexión del pie, músculos de la cara con hipomimia facial y 5/11 con ptosis palpebral. Diez presentaron voz nasal; 5, disfagia; 7 se alimentaban exclusivamente por boca; 9, hipoacusia neurosensorial o conductiva. El crecimiento fue normal, salvo en uno, y 6 presentaron osteopenia, que fue grave en dos. La CK no se normalizó hasta después de los 24 meses de TRE.
Estudiando el nivel cognitivo en la edad escolar, en 10 pacientes con EPIC que sobrevivieron con el TRE, fue normal o levemente descendido. Esto fue subestimado antes de los 5 años en relación con su compromiso motor.58
La evaluación oftalmológica pudo detectar precozmente miopía, astigmatismo y ptosis palpebral para evitar, así, la ambliopía.44
También se ha señalado que, en el sistema nervioso central (SNC), una dilatación ventricular inicial mejora con el TRE, pero, en algunos pacientes, pueden presentarse cambios en la sustancia blanca periventricular y señales anormales en los núcleos de la base y aneurismas cerebrales.40
Si bien el TRE ha mejorado la sobrevida (evidencia grado III) y la calidad de vida de la mayoría de los niños, la evolución es variable, debido a factores, como la edad de inicio del TRE, el grado del compromiso de la enfermedad, el genotipo, el estado CRIM, la presencia en la biopsia muscular de predominio de fibras y autofagia, y el alto nivel de autoanticuerpos durante el tratamiento.41,42
SEGUIMIENTO
Para evaluar la eficacia del tratamiento, es necesario un cuidadoso y reglado seguimiento multidisciplinario coordinado por un profesional con experiencia en la EP (Anexo: Ficha de seguimiento clínico. Véase en formato electrónico). La mejoría o la estabilización del paciente son indicación de continuar con el TRE.59
Agradecimientos
A los doctores Hernán Amartino, Francisco Astorino y Soledad Monges por su colaboración y sugerencias.
Anexo
Ficha de seguimiento clínico. Modificada de la referencia

1. Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Beaudet A, Scriver C, Sly W, Valle D (eds.). The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York; Mc Graw Hill, 2001:3389-420. [ Links ]
2. Pompe JC. Over idiopatische hypertrophie van het hart. Ned Tijdschr Geneeskd. 1932;76:304-11. [ Links ]
3. Hers HG. Alpha-Glucosidase deficiency in generalized glycogen-storage disease (Pompe's disease). Biochem J. 1963;86(1):11-6. [ Links ]
4. Honig J, Martiniuk F, D'Eustachio P, Zamifirescu C, et al. Confirmation of the regional localization of the genes for human acid alpha-glucosidase (GAA) and adenosine deaminase (ADA) by somatic cell hybridization. Ann Hum Genet. 1984;48(1):49-56. [ Links ]
5. Martiniuk F, Chen A, Mack A, Arvanitopoulos E, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease . Am J Med Genet. 1998;79(1):69-72. [ Links ]
6. Kishnani P, Steiner R, BaliD, Berger K, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-88. [ Links ]
7. Van den Hout HM, Hop W, Van Diggelen OP, Smeitink JA, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112(2):332-40. [ Links ]
8. Van Capelle CI, Van der Meijden JC, Van der Hout JM, Jaeken J, et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J Rare Dis. 2016;11(1):65. [ Links ]
9. Kishnani PS, Hwu WL, Mandel H, Nicolino M, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148(5):671-6. [ Links ]
10. Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet. 2012;160C(1):1-7. [ Links ]
11. Byrne BJ, Kishnani PS, Case LE, Merlini L, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab. 2011;103(1):1-11. [ Links ]
12. Laforêt P, Nicoino M, Eymard PB, Puech JP, et al. Juvenil and adult onset acid maltase deficiency in France, genotype phenotype correlation. Neurology. 2000;55(8):1122-8. [ Links ]
13. Engel AG, Gomez MR, Seybold ME, Lambert EH. The spectrum and diagnosis of acid maltase deficiency. Neurology. 1973;23(1):95-106. [ Links ]
14. Jones HN, Muller CW, Lin M, Banugaria SG, et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25(4):277-83. [ Links ]
15. Bembi B, Cerini E, Danesino C, Donati MA, et al. Management and treatment of Glycogenosis type II. Neurology. 2008;71(23 Suppl 2):S12-36. [ Links ]
16. Chen LR, Chen CA, Chiu SN, Chien YH, et al. Reversal of cardiac dysfunction after enzyme replacement in patients with infantile-oset Pompe disease. J Pediatr. 2009;155(2):271-5.e2. [ Links ]
17. Hoeksma M, Boon M, Niezen-Koning KE, Van Overbeek-van Gils L, et al. Isolated elevated serum transaminases leading to the diagnosis of asymptomatic Pompe disease. Eur J Pediatr. 2007;166(8):871-4. [ Links ]
18. Gillette PC, Nihill M, Singer DB. Electrophysiological mechanism of the short PR interval in Pompe disease. Am J Dis Child. 1974;128(5):622-6. [ Links ]
19. Barohn RJ, McVey AL, DiMauro S. Adult acid maltase deficiency. Muscle Nerve. 1993;16(6):672-6. [ Links ]
20. Alejaldre A, Diaz-Manera J, Ravaglia S, Tibaldi EC, et al. Trunk muscle involvement in late-onset Pompe disease: study of thirty patients. Neuromuscul Disord. 2012;22( Suppl 2):S148-54. [ Links ]
21. Carlier RY, Laforet P, Wary C, Mompoint D, et al. Whole-body muscle MRI in 20 patients suffering from late onset Pompe disease: Involvement patterns. Neuromuscul Disord. 2011;21(11):791-9. [ Links ]
22. Chamoles N, Niizawa G, Blanco M, Gaggiolo D, et al. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin Chim Acta. 2004;347(1-2):97-102. [ Links ]
23. Lin N, Huang J, Violante S, Orsini JJ, et al. Liquid Chromatography-tandem mass spectrometry assay of leuckocyte acid alpha-glucosidase for post newborn screening evaluation of Pompe disease. Clin Chem. 2017;63(4):842-51. [ Links ]
24. Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis. 2008;29(2-3):397-404. [ Links ]
25. Winchester B, Bali D, Bodamer O, Caillaud C, et al. Methods for a prompt and reliable laboratory diagnosis of Pompe disease: Report from an international consensus meeting. Mol Genet Metab. 2008;93(3):275-81. [ Links ]
26. Young SP, Corzo D, Kishnani P. Diagnostic value of urinay and plasma tetrasaccharides in infantile and late onset glycogen storage disease type II. Mol Genet Metab. 2005;84(3):241-2. [ Links ]
27. Taratuto AL, Dubrovsky A, Corderi J. G.P. 8.03 Misleading muscle biopsies in late onset Pompe's disease. Neuromuscul Disord. 2009;19(8-9):591-2. [ Links ]
28. Taratuto AL, Dubrovsky, Corderi J. 131. Non-Diagnostic Muscle Biopsies in Late Onset Pompe's Disease. J Neuropathol Exp Neurol. 2011;70(6):533. [ Links ]
29. Tarnopolsky M, Katzberg H, Petrof BJ, Sirrs S, et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. Can J Neurol Sci. 2016;43(4):472-85. [ Links ]
30. Erasmus MC. Pompe Center. Clinical genetics. Molecular aspects. DNA. Protein. Mutations. [Consulta: 7 de enero de 2018]. Disponible en: https://www.erasmusmc.nl/klinische_genetica/research/lijnen/pompe_center/?lang=en.
31. Kroos M, Hoogeveen-Westerveld M, Van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet. 2012;160C(1):59-68. [ Links ]
32. McIntosh P, Austin S, Sullivan J, Bailey L, et al. Three cases of multi-generational Pompe disease: Are current practices missing diagnostic and treatment opportunities? Am J Med Genet A. 2017;173(10):2628-34. [ Links ]
33. Jones HN, Crisp KD, Moss T, Strollo K, et al. Effects of respiratory muscle training in children with infantile onset Pompe disease an respiratory muscle weakness. J Pediatr Rehabil Med. 2014;7(3):255-65. [ Links ]
34. Van Capelle CI, Goedegebure A, Homans NC, Hoeve HL, et al. Hearing loss in Pompe disease revisited:results from a study of 24 children. J Inherit Metab Dis. 2010;33(5):597-602. [ Links ]
35. Case LE, Beckemeyer AA, Kishnani PS. Infantile Pompe disease on ERT: update on clinical presentation, musculoskeletal management, and excersice consideration. Am J Med Genet C Semin Med Genet. 2012:160C:69-79. [ Links ]
36. Roberts M, Kishnani PS, Van deer Ploeg AT, Müller-Felber W, et al. The prevalence and impact of scoliosis in Pompe disease: lessons learned from Pompe Registry. Mol Genet Metab. 2011;104(4):574-82. [ Links ]
37. Kishnani PS, Nicolino M, Voit T, Rogers R, et al. Chinese hamster ovary cellderived recombinant human acid alpha-glucosidase in infantileonset Pompe disease. J Pediatr. 2006;149(1):8997. [ Links ]
38. Kishnani PS, Corzo D, Nicolino M, Byrne B, et al. Recombinant human acid [alpha] glucosidase: major clinical benefits in infantileonset Pompe disease. Neurology. 2007;68(2):99-109.
39. Amartino H, Cavagnani B. Terapia de remplazo enzimático en la forma infantil de la enfermedad de Pompe; experiencia de un caso con 7 años de seguimiento en Argentina. Arch Argent Pediatr. 2012:110(4):323-7. [ Links ]
40. McIntosh PT, Hobson-Webb LD, Kazi ZB, Prater SN, et al. Neuroimaging findings in infantile Pompe patients treated with enzyme replacement therapy. Mol Genet Metab. 2018;123(2):85-91. [ Links ]
41. Nicolino M, Byrne B, Wraiht JE, Leslie N, et al. Clinical outcome after long-term treatment with alglucosidase alpha in infants and children with advanced Pompe disease. Genet Med. 2009;11(3):210-9. [ Links ]
42. Hahn A, Praetorius S, Karabul N, DieBel J, et al. Outcome of patients with classical infantile Pompe disease receiving enzyme replacement therapy in Germany. JIMD Rep. 2015;20:65-75. [ Links ]
43. Ansong AK, Li JS, Nozik-Grayck E, Ing R, et al. Electrocardiographic response to enzyme replacement therapy for Pompe disease. Genet Med. 2006;8(5):297-301. [ Links ]
44. Yang CF, Yang CC, Liao HC, Huang LY, et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J Pediatr. 2016;169:174-80.e1. [ Links ]
45. Prakalapakorn SG, Proia AD, Yanovitch TL, DeArmey S, et al. Ocular And Histologic Findings In: A Series Of Children With Infantile Pompe Disease Treated With Enzyme Replacement Therapy. J Pediatr Ophthalmol Strabismus. 2014;51(6):355-62. [ Links ]
46. Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab. 2010;99(1):26-33. [ Links ]
47. Berrier KL, Kazi ZB, Prater SN, Bali DS, et al. CRIMnegative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med. 2015;17(11):9128. [ Links ]
48. Bali DS, Goldstein JL, Banugaria S, Dai J, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Genet. 2012;160C(1):40-9. [ Links ]
49. Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe's disease. N Engl J Med. 2009;360(2):194-5. [ Links ]
50. Van Gelder CM, Poelman E, Plug I, Hoogeveen-Westerveld M, et al. Effects of a higher dose of alglucosidase alfa on ventilatorfree survival and motor outcome in classic infantile Pompe disease: an openlabel singlecenter study. J Inherit Metab Dis. 2016;39(3):383-90. [ Links ]
51. ElGharbawy AH, Mackey J, DeArmey S, Westby G, et al. An individually, modified approach to desensitize infants and young children with Pompe disease, and significant reactions to alglucosidase alfa infusions. Mol Genet Metab. 2011;104(1-2):11822. [ Links ]
52. Richard E, Douillard-Guilloux G, Caillaud C. New insights into therapeutic options for Pompe disease. IUBMB Life. 2011;63(11):979-86. [ Links ]
53. Lukas J, Pockrandt AM, Seemann S, Sharif M, et al. Enzyme enhancers for the treatment of Fabry and Pompe disease. Md Ther- 2015;23(3):456-64. [ Links ]
54. Liang Q, Stok M, Van Helsdingen Y, Van der Velden G, et al. Lentiviral Stem Cell Gene Therapy for Pompe Disease. J Neuromuscul Dis. 2015;2(s1)S64. [ Links ]
55. Wagemaker G. Lentiviral Hematopoietic Stem Cell Gene Therapy in Inherited Metabolic Disorders. Hum Gene Ther. 2014;25(10):862-5. [ Links ]
56. Porto C, Ferrara MC, Meli M, Acampora E, et al. Pharmacological Enhancement of a-glucosidase by the allosteric chaperone N-acetylcysteine. Mol Ther. 2012;20(12):2201-11. [ Links ]
57. Prater S, Banugaria S, DeArmey S, Botha EG, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med. 2012;14(9):800-10. [ Links ]
58. Ebbink BJ, Aarsen FK, Van Gelder CM, Van den Hout JM, et al. Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology. 2012;78(19):1512-8. [ Links ]
59. Pascual-Pascual SI, Nascimento A, Fernandez-Llamazares C, Medrono-Lopez C, et al. Guia Clinica de la enfermedad de Pompe Infantil. Rev Neurol. 2016;63(6):269-79. [ Links ]

