










Serviços Personalizados
Journal

Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkArchivos argentinos de pediatría
versão impressa ISSN 0325-0075versão On-line ISSN 1668-3501
Arch. argent. pediatr. vol.117 no.5 Buenos Aires out. 2019
http://dx.doi.org/10.5546/aap.2019.330
ORIGINAL ARTICLE
http://dx.doi.org/10.5546/aap.2019.eng.330
Clinical and molecular characterization of children with Noonan syndrome and other RASopathies in Argentina
Josefina Chinton, Biochemista, Victoria Huckstadt, M.D.b, Angélica Moresco, M.D.b, L. Pablo Gravina, Biochemista and M. Gabriela Obregon, M.D.b
a. Molecular Biology Laboratory, Department of Genetics.
b. Department of Genetics. Hospital de Pediatría Garrahan, Autonomous City of Buenos Aires, Argentina.
E-mail address: L. Pablo Gravina, Biochemist: pablogravina97@gmail.com
Funding: None.
Conflict of interest:
None.
Received: 7-26-2018
Accepted:2-14-2019
ABSTRACT
Introduction. RASopathies are a set of syndromes with phenotypic overlapping features caused by gene mutations involved in the RAS/MAPK pathway. They are autosomal dominantly inherited and share common clinical characteristics, including short stature, craniofacial dysmorphisms, congenital heart disease, ectodermal manifestations, and a higher risk for cancer. A molecular diagnosis is a key factor.
Objective. To identify PTPN11, SOS1, RAF1, BRAF, and HRAS mutations and compare the main clinical characteristics of patients with molecular confirmation.
Population and methods. Children with a clinical diagnosis of RASopathy assessed between August 2013 and February 2017.
Results. Mutations were identified in 71 % (87/122) of patients. The molecular test confirmed diagnosis in 73 % of patients with Noonan syndrome. The most prevalent mutation was c.922A>G (p.Asn308Asp) in the PTPN11 gene. A previously undescribed variant in RAF1 was detected: c.1467G>C (p.Leu489Phe). Cardiofaciocutaneous syndrome was confirmed in 67 % of cases with BRAF mutations. Costello syndrome and Noonan syndrome with multiple lentigines were confirmed in all cases.
Conclusion. The confirmation of clinical diagnosis allowed for a more accurate differential diagnosis. The prevalence of PTPN11 (58 %), SOS1 (10 % ), and RAF1 mutations (5 %) in children with Noonan syndrome, of PTPN11 mutations (100 %) in those with Noonan syndrome with multiple lentigines, of BRAF mutations (67 %) in those with cardiofaciocutaneous syndrome, and of HRAS mutations (100 %) in those with Costello syndrome was determined.
Key words: RASopathies; Noonan syndrome; PTPN11; RAF1; Argentina.
GLOSSARY
CFC syndrome: cardiofaciocutaneous syndrome.
CS: Costello syndrome.
HCM: hypertrophic cardiomyopathy.
NS: Noonan syndrome.
NSML: Noonan syndrome with multiple lentigines.
PS: pulmonary stenosis.
INTRODUCTION
RASopathies are a set of syndromes with phenotypic overlapping features, including Noonan syndrome (NS), Noonan syndrome with multiple lentigines (NSML), Costello syndrome (CS), cardiofaciocutaneous (CFC) syndrome, and neurofibromatosis-Noonan syndrome. RASopathies are caused mainly by gain-of-function mutations in the genes involved in the RAS/MAPK signaling pathway. This is implied in several biological functions, such as the regulation of cell proliferation and cell survival and differentiation.1
These syndromes are autosomal dominantly inherited and share some clinical characteristics.
NS (OMIM 163950) is the most common one in this set, with an incidence of 1:1000 to 1:2500 live births.1 Its main characteristics include short stature, congenital heart disease, craniofacial dysmorphisms, and thoracic malformations.2 Sometimes, intellectual disability, hearing loss, short neck, clotting alterations, and cryptorchidism (in males) may occur.3 Children with NS have a higher risk for developing different types of cancer.4 NS is caused by the following gene mutations: PTPN11 (50 %), SOS1 (10-15 %), RAF1 (5-15 %), RIT1 (4-9 %), KRAS (<5 %), NRAS (<1 %), BRAF (<1 %), SHOC2 (<1 %), MEK1 (<1 %), and CBL (<1 %).5-9 Approximately 70-80 % of patients with NS have these gene mutations.10 The causative mutations have not been identified in the other 20-30 %.11De novo mutations account for 60 % of NS cases.12
The clinical characteristics of NSML (previously known as LEOPARD syndrome, OMIM 151100) include the presence of multiple lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis (PS), genital abnormalities, delayed growth, and hearing loss. It is caused by PTPN11 (90 %), RAF1 (5 %), and BRAF (5 %) mutations.6
CFC syndrome (OMIM 115150) is characterized by craniofacial dysmorphisms, neurological development delay, congenital heart disease, and ectodermal and musculoskeletal manifestations. It is caused by de novo mutations in the BRAF gene (75 %) and the MEK1/MEK2 gene (25 %). KRAS mutations were reported in a lower percentage of cases (<1 %).13,14
CS (OMIM 218040) is one of the less common RASopathies. Its characteristics include intellectual disability, high birth weight, difficulty sucking in the neonatal period, perioral and perianal papillomas, and deep palmar and plantar creases. Patients with CS have a 13 % probability of developing tumors.15It is caused by heterozygous mutations in the HRAS protooncogene in more than 80 % of cases.16
Given the overlapping clinical features of these syndromes and their variable expression, molecular testing is critical to establish an accurate diagnosis. The objective of this study was to identify PTPN11, SOS1, RAF1, BRAF, and HRAS mutations in children with RASopathies and compare the main clinical characteristics of patients with molecular confirmation.
POPULATION AND METHODS
This was a cross-sectional, observational, and descriptive study that included patients with a clinical diagnosis of RASopathy2,917 assessed between August 2013 and February 2017 by the Department of Genetics of Hospital de Pediatría Garrahan. Two clinical geneticists recorded the presence of dysmorphisms, congenital heart disease, skeletal and kidney abnormalities, ectodermal manifestations, cryptorchidism, hearing loss, clotting alterations, splenomegaly, lymphatic dysplasia, intellectual disability, and cancer.
Anthropometric parameters were analyzed by sex and age, and are described as standard deviation (SD). The Argentine weight and height charts were used as the reference population,18 and the Nellhaus chart, for head circumference.19
A written informed consent was approved by the hospital's Ethics Committee and obtained from patients and/or their parents.
Molecular analysis
Deoxyribonucleic acid (DNA) samples from peripheral blood lymphocytes were analyzed for mutations using the Sanger sequencing technique (ABI BigDye Terminator Sequencing Kit V1.1; Applied Biosystems) with an automated capillary sequencer (ABI 3130, Applied Biosystems). Exons 2, 3, 4, 7, 8, 12, and 13 of the PTPN11 gene, exons 6 and 10 of the SOS1 gene, exons 7, 14, and 17 of the RAF1 gene, exons 6, 12, and 15 of the BRAF gene, and exon 2 of the HRAS gene were analyzed. Sequencing was done by stages, in order of relevance, according to the mutation frequency described in the main exons of each gene.6,9,13,14,20 The HRAS gene was tested in patients with suspected CS.16
Statistical analysis
Fisher's exact test was done. A p value < 0.05 was considered statistically significant.
RESULTS
A total of 122 patients (56 females and 66 males) diagnosed with RASopathy were assessed. The median age at the time of clinical diagnosis was 6 years (range: 0-19 years). Most patients were of European descent, chiefly Spanish and Italian. Mutations were detected in 87 patients (71 %). Initially, 100 patients received a clinical diagnosis of NS; 16, CFC syndrome; 3, NSML; and 3, CS.
The mutation identified in 3 patients led to a modification in the initial diagnosis. One of them was assessed at 2 years old due to dysmorphisms, short stature, and ectodermal manifestations and received an initial diagnosis of CFC syndrome. This patient was reassessed at 16 years old and showed no intellectual disability or heart disease. A new variant in the RAF1 gene was detected and the diagnosis was changed to NS. In another 2-year-old patient without skin manifestations at the time of assessment and with an initial diagnosis of NS, the p.Thr468Met mutation in the PTPN11 gene was detected, which had been previously associated with NSML; the subsequent assessment of his father showed multiple lentigines at the time of the physical examination and this changed the diagnosis to NSML. Finally, there was a patient with a clinical diagnosis of CS, but the detection of a BRAF mutation led to changing it to CFC syndrome.
In addition, a patient with clinical diagnosis of CFC syndrome due to ectodermal manifestations, such as keratosis pilaris, curly hair, and sparse eyebrows, without cognitive deficit or congenital heart disease, had the c.806C>T mutation (p.Met269Thr) in the SOS1 gene, which is rarely associated with CFC syndrome.
In five patients with a negative molecular test, follow-up allowed to establish a diagnosis other than RASopathy. The clinical reassessment determined a total of 96 patients with a diagnosis of NS; 15, CFC syndrome; 4, NSML; and 2, CS.
The molecular test confirmed the diagnosis in 71/96 patients with NS (73 %); 56 (58 %) had PTPN11 mutations; 10, (10 %) SOS1 mutations; and 5 (5 %), RAF1 mutations. No mutations were detected with the methodology used here in the other 25 patients. All patients with NSML had PTPN11 mutations. CFC syndrome was confirmed in 10/15 cases with BRAF mutations. CS was confirmed in 2 patients with HRAS mutations.
Among patients in whom a mutation was detected, 72 corresponded to sporadic cases and 15, to familial cases; transmission was maternal in 11 of them.
The clinical characteristics are summarized in Table 1. The 96 patients with NS had facial dysmorphisms; 51 % of them had short stature, which was more common among those with PTPN11 mutations. Also, 76 % of patients had heart disease, with PS as the most frequent condition. Hypertrophic cardiomyopathy (HCM), aortic stenosis, tetralogy of Fallot, atrial septal defect, and ventricular septal defect were also detected.
Table 1. Clinical characteristics of patients with RASopathies

The comparison of the main phenotypic features of patients with molecular confirmation of NS and NSML (Table 2) showed that the latter had a strong association with HCM, a smaller presence of short stature and overall developmental delay or intellectual disability (p < 0.05). Skin manifestations were observed in 75 % of patients with NSML, but no significant differences were observed in terms of this clinical characteristic when compared to NS patients (Table 2).
Table 2. Comparison of the main clinical characteristics of patients with Noonan syndrome, cardiofaciocutaneous syndrome, and Noonan syndrome with multiple lentigines

A strong relation between patients with CFC syndrome and ectodermal manifestations and hearing loss was observed in comparison with those who had NS (p < 0.05) (Table 2).
No significant differences were seen in relation to ectodermal manifestations, short stature, and overall developmental delay or intellectual disability when comparing clinical manifestations between NS patients with PTPN11 and SOS1 mutations. NS patients with RAF1 mutations had a higher incidence of HCM compared to those with PTPN11 mutations (p = 0.015).
Molecular characteristics and distribution of mutations
The greatest number of mutations was detected in the PTPN11 gene (60), followed by the SOS1 (10) and BRAF (10) genes (Table 3). RAF1 and HRAS mutations were detected in 5 and 2 patients, respectively. All the mutations that were detected had been previously reported in the bibliography, except for a new variant in the RAF1 gene, c.1467G>C (p.Leu489Phe). It was registered in the Leiden Open Variation Database (LOVD).21
Table 3. Mutations detected in patients with RASopathy
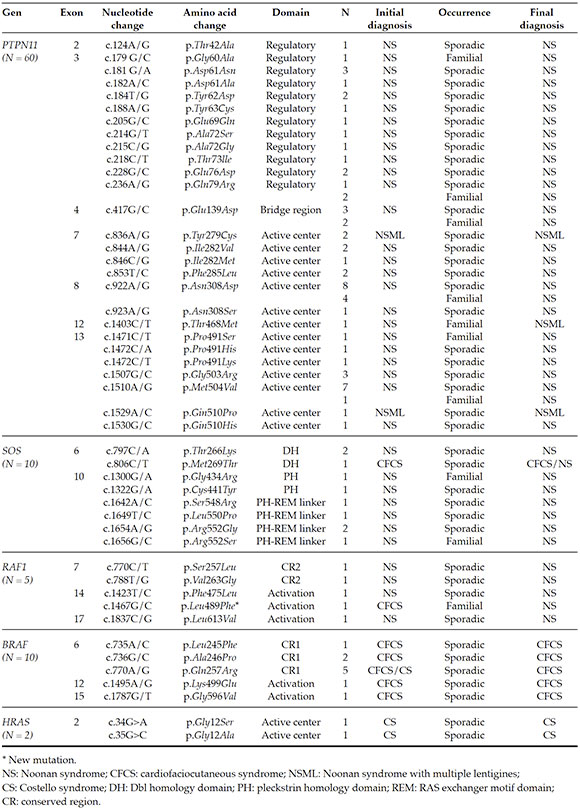
Familial cases were observed in 12 patients with a mutation in the PTPN11 gene, 2 in the SOS1 gene, and 1 in the RAF1 gene (Table 3). Also, 75 % of mutations detected in the PTPN11 gene were located in exons 3, 8, and 13.
In silico analyses were done to predict the functional effect of the new variant detected in the RAF1 gene, c.1467G>C. The variant was analyzed using different biocomputing tools that assessed the functional effects of singlenucleotide variants in human beings (Mutation Taster, PolyPhen2, and SIFT). The variant was identified as probably deleterious (score: 0.997) with Polyphen; pathogenic, with Mutation Taster; and deleterious (score: -3.605), with SIFT. This variant had been previously detected in the skin of a patient with malignant melanoma (COSM5398071) and has been described in the COSMIC database (Catalogue of Somatic Mutations in Cancer).
DISCUSSION
In this study, mutations were detected in 71 % of patients with NS and other RASopathies. The most prevalent mutation in our cohort was p.Asn308Asp, in the PTPN11 gene. This is also one of the most prevalent mutations in the European population,20,22 but it has not been reported in the Chilean or Brazilian population.23,24 This may be due to the descent of Argentine patients. The ethnic origin of our population is the result of mixed native genes and genes from, mostly, Europe's Mediterranean countries, especially Spain and Italy and, to a lesser extent, Central and Eastern Europe and Middle East.25
In relation to NS, the molecular test confirmed the clinical diagnosis in 73 % of cases, which is consistent with the bibliography.26 In this cohort, only 15 % of patients corresponded to familial cases, lower than what has been published by Tartaglia et al., who reported that 30-75 % of NS cases were familial.27 This may be because not all parents were available for the clinical assessment and the genetic analysis.
Patients with NS and PTPN11 or SOS1 mutations typically have a high incidence of PS and a lower prevalence of HCM.5 In our study, similar results were reported: 68 % of patients with PTPN11 mutations had PS; and 2 %, HCM. Among patients with SOS1 mutations, 90 % had a heart disease, with PS being the most prevalent one.
SOS1 mutations are associated with a phenotype within the clinical spectrum of NS, but it is characterized by a high prevalence of ectodermal manifestations, a low incidence of cognitive deficit, and short stature.14 In our cohort, ectodermal manifestations were present in 50 % of NS patients with SOS1 mutations; most of them had sparse eyebrows. Short stature and intellectual disability were observed in 40 % of patients. This may not have been similar to what has been reported in the bibliography14 due to the relatively small number of patients with SOS1 mutations and the young age of assessed patients.
RAF1 mutations have been associated with HCM, arrhythmia, and intellectual disability.6 Some previous publications have proposed that approximately 85 % of patients with RAF1 mutations tend to develop HCM.28 For this reason, it is very important that these patients receive an intensive cardiac follow-up. A previously undescribed variant in this gene, c.1467G>C (p.Leu489Phe), was detected, in which both the in silico analysis and the patient's and mother's clinical characteristics help to link it to the syndrome.
As described in the bibliography, patients with RASopathies typically have a greater risk for tumor development. This characteristic was also observed in our cohort, and 3 NS patients who had PTPN11 mutations developed acute lymphoblastic leukemia and paravertebral ganglioneuroma.
In NSML, the main clinical characteristics include skin manifestations, which sometimes do not appear until after puberty. In our cohort, only 1 patient did not have skin manifestations but he was only 2 years old at the time of assessment.
Among patients with a clinical diagnosis of CFC syndrome, the molecular test confirmed diagnosis in 67 % of cases, which is consistent with previous publications.13These patients had a high rate of intellectual disability and ectodermal manifestations. These two characteristics were also prevalent among the patients in our cohort. This study found a high incidence of hearing loss (40 %), which, although it was not a distinguishing feature of CFC syndrome, had already been reported by Carcavillaet al.29
As mentioned in the results, in one of our patients diagnosed with CFC syndrome, a mutation was identified in the SOS1 gene: c.806C>T (p.Met269Thr). Although this gene was not associated with CFC syndrome, Tartaglia et al.30 reported on patients with the CFC syndrome phenotype who had SOS1 mutations and proposed that the clinical characteristics of NS and CFC syndrome overlapped. Our case supported the observations of those authors.
The Sanger sequencing technique, which was used in this study and is available in our hospital, did not detect mutations in 29 % of cases. Although this is a sensitive and specific methodology, it is a tedious, slow, and costly method to study genetically heterogeneous syndromes. Therefore, a next generation sequencing (NGS) panel focused on a set of RASopathy-associated genes is a more adequate and cost-effective option to diagnose these syndromes.
To sum up, this study determined, in Argentine children, the prevalence of mutations in the following genes: PTPN11 (58 %), SOS1 (10 %), and RAF1 (5 %) in NS; PTPN11 (100 %) in NSML; BRAF (67 %) in CFC syndrome; and HRAS (100 %) in CS. Given the overlapping clinical characteristics of these syndromes, especially during childhood, molecular testing is a critical tool for diagnostic confirmation. In addition, it helps to provide more accurate guidance on prognosis, the adequate management of specific complications, e.g., cardiac, hematological or oncological, the risk for recurrence, and the analysis of other family members at risk.
Acknowledgments
We are particularly grateful to the patients and their families for participating in this study, the physicians in other sites for referring their patients to our hospital, and all the health care providers and technicians working at Hospital Garrahan.
1. Tartaglia M1, Kalidas K, Shaw A, Song X, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002; 70(6):1555-63. [ Links ]
2. Van der Burgt I, Berends E, Lommen E, Van Beersum S, et al. Clinical and molecular studies in a large Dutch family with Noonan Syndrome. Am JMed Genet. 1994; 53(5)187-91. [ Links ]
3. Kosaki K, Suzuki T, Muroya K, Hasegawa T, et al. PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome. J Clin Endocrinol Metab.2002; 87(8):3529-33. [ Links ]
4. Villani A, Greer MC, Kalish JM, Nakagawara A, et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin Cancer Res. 2017; 23(12):e83-90. [ Links ]
5. Tartaglia M, Mehler EL, Goldberg R, Zampino G, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet.2001; 29(4);465-8. [ Links ]
6. Pandit B, Sarkozy A, Pennacchio LA, Carta C, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet.2007; 39(8):1007-12. [ Links ]
7. Roberts AE, Araki T, Swanson KD, Montgomery KT, et al. Germline gain-of function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007; 39(1):70-4. [ Links ]
8. Allanson JE. The clinical phenotype of Noonan syndrome. In: Zenker M (ed.). Noonan Syndrome and Related Disorders. Monogr Hum Genet. Basel: Karger, 2009; 17:9-19. [ Links ]
9. Sarkozy A, Digilio MC, Zampino G, Dallapiccola B, et al. Leopard syndrome: clinical aspects and molecular pathogenesis. In: Zenker M (ed.). Noonan Syndrome and Related Disorders - A Matter of Deregulated Ras Signaling. Monogr Hum Genet. Basel: Karger, 2009; 17:55-65. [ Links ]
10. Hernández-Porras I, Guerra C. Modeling RASopathies with Genetically Modified Mouse Models. Methods Mol Biol. 2017; 1487:379-408. [ Links ]
11. Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016; 61(1):33-9. [ Links ]
12. Shaw AC, Kalidas K, Crosby AH, Jeffery S, et al. The natural history of Noonan syndrome a long term-follow up study. Arch Dis Child. 2007; 92(2):128-32. [ Links ]
13. Sarkozy A, Carta C, Moretti S, Zampino G, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009; 30(4):695-702. [ Links ]
14. Lepri F, De Luca A, Stella L, Rossi C, et al. SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat. 2011; 32(7):760-72. [ Links ]
15. Niihori T, Aoki Y, Okamoto N, Kurosawa K, et al. HRAS mutants identified in Costello syndrome patients can induce cellular senescence: possible implications for the pathogenesis of Costello syndrome. J Hum Genet. 2011; 56(10):707-15. [ Links ]
16. Aoki Y, Niihori T, Kawame H, Kurosawa K, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005; 37(10):1038-40. [ Links ]
17. Roberts A, Allanson J, Jadico SK, Kavamura MI, et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006; 43(11):833-42. [ Links ]
18. Lejarraga H, Del Pino M, Fano V, et al. Referencias de peso y estatura desde el nacimiento hasta la madurez para niñas y niños argentinos. Incorporación de datos de la OMS de 0 a 2 años, recalculo de percentilos para la obtención de valores LMS. Arch Argent Pediatr. 2009; 107(2):126-33. [ Links ]
19. Nelhaus G. Head circumference from birth to eighteen years: practical composite international and interracial graphs. Pediatrics.1968; 41(1):106-14. [ Links ]
20. Carcavilla A, Santomé JL, Galbis L, Ezquieta B. Síndrome de Noonan. Rev Esp Endocrinol Pediatr. 2013; 4(Suppl):71-85. [ Links ]
21. LOVD. Leiden Open Variation Database. Leiden (Netherlands): Leiden University Medical Center; 2004. [Consulta: 15 de febrero de 2019]. Disponible en: www.lovd.nI/3.0/home.
22. Cizmárová M, Hlinková K, Bertok S, Krotnik P, et al. New Mutations Associated with Rasopathies in a Central European Population and Genotype-Phenotype Correlations. Ann Hum Genet. 2016; 80(l):50-62. [ Links ]
23. Rodríguez FA, Unanue N, Hernández MI, Heath KE, et al. Molecular characterization of Chilean patients with a clinical diagnosis of Noonan syndrome. J Pediatr Endocrinol Metab. 2014; 27(3-4):305-9. [ Links ]
24. Bertola DR, Pereira AC, Albano LM, De Oliveira PS, et al. PTPN11 gene analysis in 74 Brazilian patients with Noonan syndrome or Noonan-like phenotype. Genet Test. 2006; 10(3):186-91. [ Links ]
25. Maccio GA, Elizalde D. La población no nativa de la Argentina. Buenos Aires: Instituto Nacional de Estadisticas y Censos; 1996. [ Links ]
26. Tafazoli A, Eshraghi P, Pantaleoni F, Vakili R, et al. Novel mutations and their genotype-phenotype correlations in patients with Noonan syndrome, using next generation sequencing. Adv Med Sci. 2018; 63(1):87-93. [ Links ]
27. Tartaglia M, Cordeddu V, Chang H, Shaw A, et al. Paternal germline origin and sex-ratio distortion in transmission of PTPN11 mutations in Noonan syndrome. Am J Hum Genet.2004; 75(3):492-7. [ Links ]
28. Gelb BD, Roberts AE, Tartaglia M. Cardiomyopathies in Noonan syndrome and the other RASopathies. Prog Pediatr Cardiol. 2015; 39(1):13-9. [ Links ]
29. Carcavilla A, García-Miñaúr S, Pérez-Aytés A, Vendrell T, et al. Síndrome cardiofaciocutáneo, un trastorno relacionado con el síndrome de Noonan: hallazgos clínicos y moleculares en 11 pacientes. Med Clin (Barc). 2015; 144(2):67-72. [ Links ]
30. Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007; 39(1):75-9. [ Links ]

