










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. v.39 n.3 La Plata jun./sept. 2005
HEMATOLOGÍA
Homeostasis del hierro.
Mecanismos de absorción, captación celular y regulación*
Gladys Pérez1, Daniela Vittori1, Nicolás Pregi2, Graciela Garbossa3, Alcira Nesse3
1. Doctor en Química Biológica.
2. Licenciado en Ciencias Biológicas.
3. Doctora en Ciencias Químicas.
* Laboratorio de Análisis Biológicos, Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires.
Resumen
El hierro es un elemento esencial para los organismos vivientes. En soluciones acuosas puede encontrarse en dos estados de oxidación estables: ferroso (Fe2+) y férrico (Fe3+), propiedad que le permite participar en reacciones que abarcan gran parte de la bioquímica. En el organismo, el hierro se encuentra formando parte de dos compartimientos: uno funcional y otro de depósito. El transporte de hierro unido a transferrina permite el intercambio del metal entre ambos compartimientos mientras que su exceso se deposita intracelularmente como ferritina o hemosiderina. En condiciones fisiológicas, el hierro se encuentra asociado a una proteína ya que su presencia en forma aislada puede producir daños graves en los tejidos. Esta toxicidad se debe a la habilidad del hierro libre de generar especies reactivas de oxígeno. Dado que tanto la deficiencia como la sobrecarga de este metal esencial son perjudiciales, su absorción, concentración y estado redox deben ser regulados cuidadosamente. En este trabajo se revisan las vías que median la absorción intestinal y la incorporación celular de hierro y se detallan los diferentes mecanismos que participan en su homeostasis. Finalmente, y considerando que el espectro de los desórdenes conocidos del metabolismo del hierro se ha expandido en los últimos años, se describen algunas de las alteraciones más comunes.
Palabras clave: hierro * transferrina * receptores de transferrina * vías alternativas de captación de hierro * hepcidina * anemia * hemocromatosis
Summary
Iron homeostasis. Mechanisms of absorption, uptake, and regulation
Iron is an essential nutrient required by every living being. This transition metal is involved in the majority of the metabolic processes, partly because of its ability to reversibly and readily cycle between ferrous (Fe2+) and ferric (Fe3+) oxidation states. Iron must be transported and stored as a component of protein complexes. Ferric ions circulate bound to plasma protein transferrin and accumulate within cells in the form of ferritin or hemosiderin. This is due to the fact that iron as a free cation can participate in a number of reactions to produce free radical species, which in turn can severely damage organs. Taking into account that both iron deficiency and iron overload are deleterious, a strict balance between iron absorption and utilization is required. In this work, the distinct pathways of gastrointestinal absorption and cellular uptake of iron are reviewed, giving details of the mechanisms involved in iron homeostasis under different conditions. Condition of iron deficiency and overload are discussed in terms of mechanisms leading to restoration of iron homeostasis.
Key words: iron * transferrin * transferrin receptors * non-transferrin bound iron uptake * hepcidin * anemia * hemochromatosis
INTRODUCCIÓN
El hierro es un elemento esencial para todos los organismos vivientes. En la primera etapa de la evolución de la vida, los seres primitivos lo utilizaban como parte de su sistema de generación de energía, existiendo en su estado ferroso debido al escaso oxígeno ambiental. Cuando la concentración de oxígeno aumentó, predominaron las formas oxidadas, poco solubles y los organismos tuvieron que sintetizar moléculas que tuvieran capacidad de unir hierro para poder así aprovechar mejor su presencia. En los animales vertebrados esta función es realizada por las proteínas transferrina (Tf) y ferritina, quienes son, respectivamente, las responsables del transporte y almacenamiento de hierro (1). Estas proteínas unen el metal muy estrechamente para evitar la formación de productos hidrolíticos insolubles, pero como el proceso es reversible, el elemento se encuentra disponible ante la demanda celular. La combinación del hierro con una proteína evita, además, su pérdida vía filtración glomerular.
En soluciones acuosas, el hierro puede encontrarse en dos estados de oxidación estables: Fe2+ (ferroso) y Fe3+ (férrico). Esta propiedad lo hace capaz de participar en reacciones que abarcan gran parte de la bioquímica, incluyendo aquéllas que controlan el flujo de electrones a través de rutas bioenergéticas, la síntesis de ADN y el aporte de oxígeno a los tejidos (1). Entre las hemoproteínas que utilizan hierro como cofactor se encuentran las que participan en el metabolismo del oxígeno (oxidasas, peroxidasas, catalasas e hidroxilasas), en la transferencia de electrones (citocromos) y en el transporte de oxígeno (hemoglobina) (2).
El hierro en el organismo se encuentra formando parte de dos compartimientos: uno funcional, que incluye los diversos compuestos celulares que contienen o requieren hierro, y otro de depósito, el cual constituye la reserva corporal del metal. El transporte de hierro unido a la Tf plasmática facilita el intercambio del metal entre ambos compartimientos. El exceso de hierro se deposita intracelularmente asociado a ferritina y hemosiderina, fundamentalmente en el sistema monocito-macrófago del bazo, el hígado y la médula ósea.
En condiciones fisiológicas el hierro está unido a una proteína ya que su presencia aislada, como en los casos de intoxicación por el metal, produce daños graves en los tejidos. Esta toxicidad se debe a la habilidad del hierro libre de generar, en conjunción con el oxígeno, radicales hidroxilos que pueden causar peroxidación de las membranas lipídicas y otros constituyentes celulares (3). Por esta razón, la absorción, concentración y estado redox de este metal, deben ser regulados cuidadosamente: si la cantidad de hierro presente es escasa, se produce anemia y si se encuentra en exceso, causa daño en los órganos por siderosis.
VÍAS DE ABSORCIÓN INTESTINAL DE HIERRO
La circulación del hierro entre los compartimientos de depósito y utilización constituye un ciclo muy eficiente y prácticamente cerrado. Dado que sólo una pequeña proporción del metal es excretada, la necesidad diaria de incorporación de hierro en un individuo es muy baja. Por lo tanto, sólo una pequeña proporción del total del metal ingerido es absorbida (aproximadamente el 10%).
Aunque el hierro puede ser absorbido a lo largo de todo el intestino, este proceso es más eficiente en el duodeno.
El hierro dietario se encuentra principalmente en estado férrico o como hierro hémico, mientras que el incorporado a través de productos farmacológicos usualmente está presente como sal ferrosa. El Fe3+ es insoluble en soluciones con pH mayor a 3, por lo que, en el estómago, se forman complejos solubles del metal que aumentan su disponibilidad para ser absorbido en el duodeno. Por otra parte, en el lumen del intestino se forman cantidades variables de iones ferrosos como consecuencia de la reducción del hierro férrico por agentes dietarios (por ejemplo, ácido ascórbico). En consecuencia, ambos iones (ferroso y férrico) pueden presentarse ante las células intestinales.
Los diferentes mecanismos descriptos para la absorción de hierro, esquematizados en la Figura 1, se resumen a continuación:
• Los iones férricos pueden ser absorbidos vía una proteína de membrana miembro de la familia de las integrinas, la b3-integrina. Luego, son transferidos a la proteína chaperona mobilferrina (4).
• La absorción de los iones ferrosos es facilitada por el transportador de metales divalentes DMT1 (divalent metal transporter 1), también conocido como DCT1 (divalent cation transporter 1) o Nramp2 (natural-resistance-associated macrophage protein 2). Por otra parte, la proteína DcytB (duodenal cytochrome b), presente en la superficie apical del enterocito, reduce los iones férricos dietarios, los cuales pueden entonces ser incorporados también vía DMT1 (5). La importancia de esta última proteína en la absorción intestinal de hierro ha sido demostrada en estudios genéticos en ratas Belgrade portadoras de una mutación en el locus DMT1. Estos animales exhiben anemia microcítica e hipocrómica severa debido a un defecto en la absorción intestinal de hierro así como también en la captación y utilización del metal en los tejidos periféricos, incluyendo los precursores eritroides (6).
• El hemo es liberado de mioglobina y hemoglobina como consecuencia de la digestión proteolítica llevada a cabo por enzimas pancreáticas. Posteriormente, es incorporado por las células absortivas del intestino delgado como una metaloporfirina intacta (7). El proceso de transporte es mediado por una proteína específica localizada en la cara apical de la membrana del enterocito (8). Dentro de la célula, el hemo es degradado por la hemooxigenasa, liberándose de esta manera el hierro inorgánico de la estructura tetrapirrólica.
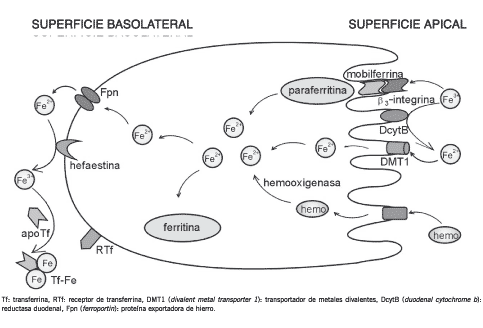
Figura 1. Mecanismos de absorción intestinal de hierro.
Tf: transferrina, RTf: receptor de transferrina, DMT1 (divalent metal transporter 1): transportador de metales divalentes, DcytB (duodenal cytochrome b): reductasa duodenal, Fpn (ferroportin): proteína exportadora de hierro.
Una vez en el interior del enterocito, el metal absorbido a través de cualquiera de las vías descriptas es convertido a su estado ferroso, paso que es realizado por un gran complejo proteico citoplasmático llamado paraferritina. El mismo incluye proteínas como b3-integrina, mobilferrina, flavin monooxigenasa y b2-microglobulina y utiliza una cadena de transporte de electrones con energía derivada de NADPH para llevar a cabo la reducción del hierro absorbido (9).
Posteriormente, los iones ferrosos pueden ser almacenados en la ferritina o alcanzar la membrana basolateral del enterocito donde son conducidos por la proteína transportadora transmembrana ferroportina (Fpn), también llamada Ireg1 (iron-regulated transporter 1) o MTP1 (metal transporter protein 1). La proteína de membrana hefaestina o la ceruloplasmina plasmática promueven la oxidación del hierro facilitando de esta manera su incorporación a la apotransferrina circulante (10).
Debido a que no existe una vía fisiológica para la excreción de este metal esencial, su absorción a nivel duodenal está cuidadosamente regulada para mantener un equilibrio entre la incorporación y la pérdida corporal. La membrana basolateral del enterocito expresa receptores para Tf que permiten la entrada del hierro transportado por esta proteína. El metal incorporado en este proceso «informa» a la célula sobre el estatus férrico del organismo, induciendo la regulación negativa de su captación vía DMT1 e integrina-mobilferrina (11).
Sin embargo, parece que el transportador basolateral Fpn sería el principal punto de regulación de la absorción de hierro dietario en respuesta a los requerimientos sistémicos, mientras que la regulación del transporte apical serviría de mecanismo de seguridad (12). La hepcidina, un pequeño péptido antimicrobiano producido por el hígado, se perfila como la principal responsable de esta regulación. De acuerdo con este modelo, la producción hepática de hepcidina estaría regulada por el grado de saturación de la Tf y el nivel de receptores para esta proteína a nivel hepático (RTf y RTf2), de modo que cuando la relación Tf-diférrica/RTf aumenta, se induce la secreción de hepcidina. La unión de esta hormona proteica a la Fpn induce la internalización y posterior degradación de la proteína transportadora. Como consecuencia de la disminución de la exportación de hierro, se produce la inhibición de su adquisición por parte de la Tf plasmática y el aumento de la concentración del metal en el enterocito que, a su vez, conduciría a una inhibición de su transporte apical.
Por el contrario, cuando la relación Tf-diférrica/ RTf disminuye, cesa la producción hepática de hepcidina y se restaura la absorción de hierro.
La regulación post-traduccional de la Fpn por hepcidina completa un círculo homeostático: el hierro regula la secreción de hepcidina, la cual a su vez, controla la concentración de Fpn en la superficie basolateral de los enterocitos (12-14)
TRANSPORTE DE HIERRO
La concentración de hierro en plasma de individuos adultos normales oscila alrededor de 1,5 µg/mL. Está predominantemente unido a Tf y en pequeña proporción a albúmina o a especies de bajo peso molecular. La cantidad de hierro requerida diariamente es captada por la Tf en las células del lumen intestinal y en los lugares de degradación de la hemoglobina (sistema monocito-macrófago) (11). Esta proteína transporta y cede el catión en los sitios de síntesis de hemoglobina y de enzimas que contienen hierro.
La Tf sérica o siderofilina es una proteína plasmática que se halla también en otros fuidos de los vertebrados, tales como líquido cefalorraquídeo, leche y semen. Es una glicoproteína de aproximadamente 80 kDa, constituida por una única cadena polipeptídica a la que se unen hidratos de carbono formando dos ramas idénticas y casi simétricas. Su ligando natural es el hierro trivalente al que une con alta afinidad. En condiciones fisiológicas la constante de estabilidad efectiva es del orden de 1020 M-1 (15)(16). Además, esta proteína puede unir una variedad de iones multivalentes (1)(15)(16).
La Tf es capaz de unir un ion férrico a cada uno de sus dos sitios, los cuales poseen 40% de homología pero difieren en sus propiedades químicas, cinéticas, espectroscópicas y termodinámicas, siendo el dominio de mayor afinidad el ubicado en la región C-terminal (17).
Para que la unión con hierro pueda establecerse, se requiere la unión simultánea de un anión. Por cada ion férrico que se incorpora se liga concomitantemente un anión carbonato o bicarbonato y se liberan, aproximadamente, tres protones (16). La existencia de una cooperatividad recíproca entre la unión del ion metálico y la del anión indica que la Tf tiene un estricto requerimiento del anión sinergista para facilitar la unión con el hierro. La función del anión sería la de actuar como ligando «reforzador» de la unión entre la proteína y el metal. A pH fisiológico, en presencia de ion bicarbonato y en ausencia de agentes complejantes, el hierro se une a cualquiera de los dos sitios de la Tf humana. Luego, factores cinéticos gobiernan la distribución del metal entre los dos sitios (1).
Normalmente, 70% de la Tf plasmática no está saturada con el metal esencial y, por lo tanto, la reserva total de esta proteína actúa como amortiguador frente a grandes cantidades de hierro absorbido o liberado, las que, de otra manera, resultarían tóxicas cuando el metal está libre en su estado oxidado. En situaciones de saturación de la capacidad de transporte de Tf, el hierro puede unirse a otros ligandos, como citrato, constituyendo el llamado pool de hierro no unido a Tf (NTBI, non-transferrin bound iron).
Además de la Tf sérica, existen otros dos tipos de transferrinas: la lactoferrina o lactotransferrina, presente en algunas secreciones de los mamíferos (leche, lágrimas, jugo pancreático) y en contenidos intracelulares de leucocitos y la ovotransferrina, presente en las secreciones de los oviductos de aves y reptiles y en la clara de huevo. Estos miembros de la familia desempeñan un rol importante en la defensa contra las infecciones, restringiendo la disponibilidad de hierro para el metabolismo microbiano (18).
EL RECEPTOR DE TRANSFERRINA Y LA INCORPORACIÓN CELULAR DE HIERRO
El número y estabilidad de los receptores de transferrina (RTf) en la superficie celular son los primeros determinantes de la captación de hierro. Estos receptores no sólo son importantes para facilitar el acceso del metal esencial a la célula sino que cumplen, además, un rol crítico en la liberación del hierro del complejo con Tf en el interior celular.
El RTf es una glicoproteína homodimérica transmembrana de aproximadamente 190 kDa. Los monómeros se encuentran ligados entre sí por dos puentes disulfuro y cada molécula de RTf posee capacidad para unir dos moléculas de Tf (19).
Virtualmente, todas las células de los mamíferos poseen RTf en su superficie, pero éstos se expresan en mayor número en células hepáticas, de placenta y en progenitores eritroides (20). El número de receptores es aproximadamente constante en células en reposo, pero aumenta marcadamente durante la proliferación celular.
El hierro transportado por Tf ingresa a las células a través de un proceso de endocitosis mediada por RTf. El primer paso es la unión de Tf al receptor en la membrana. A pH 7,4 el RTf posee muy baja afinidad por la apoTf, intermedia por la Tf monoférrica y cuatro veces más elevada por la Tf diférrica, estimada esta última en 2-7¥10-9 M (21). El cambio de cargas resultante de la unión del hierro y el bicarbonato con liberación de protones, estaría relacionado con esta afinidad diferencial de los receptores por la Tf según su grado de saturación con hierro.
Los RTf se encuentran concentrados en fosas (invaginaciones de la membrana plasmática) revestidas internamente por la proteína clatrina. Una vez formado el complejo Tf-RTf, el proceso de invaginación se completa, dando origen a una vesícula revestida que es transportada junto con el complejo ligando-receptor al interior celular. Una vez que la clatrina es removida, la vesícula resultante se fusiona con un endosoma (22) (Figura 2).
El pH del interior del compartimiento endosomal oscila alrededor de 5,5 debido a la acción de una bomba de protones dependiente de ATP presente en su membrana, la cual bombea protones desde el citosol al interior del endosoma. A este pH, el hierro es liberado del complejo Tf-RTf como ion férrico. La unión cooperativa metal-bicarbonato no sólo es importante para la asociación del hierro a la Tf sino también, para su liberación. La unión anión-Tf, relativamente lábil, parece proveer un mecanismo para la remoción fisiológica del hierro. El carbonato se desaloja primero y, por ello, la unión metal-Tf es fácilmente desestabilizada, produciéndose la liberación del hierro mientras que la proteína permanece intacta (1). Posteriormente, el metal es reducido por una ferri-reductasa endosomal y transportado al citoplasma por el transportador de cationes divalentes DMT1 (DCT1, Nramp2) (6)(23).
En contraste con la mayoría de los ligandos que se disocian de su receptor y entran en una ruta de fusión con lisosoma y degradación, la elevada afinidad del RTf por apoTf a pH ácido los mantiene unidos (24). El complejo apoTf-RTf es transportado intacto hacia la membrana plasmática donde, al tomar contacto nuevamente con el medio extracelular de pH neutro, se disocia. La apoproteína queda entonces disponible para captar nuevamente hierro y comenzar otro ciclo de transporte e internalización.
Una vez en el citoplasma, el ion ferroso incorporado puede seguir tres destinos:
• pool de utilización, es decir, las proteínas celulares que requieren hierro
• pool de almacenamiento, constituido primariamente por ferritina y hemosiderina
• pool regulatorio, el cual incluye a las proteínas encargadas de detectar variaciones en los niveles intracelulares del metal.
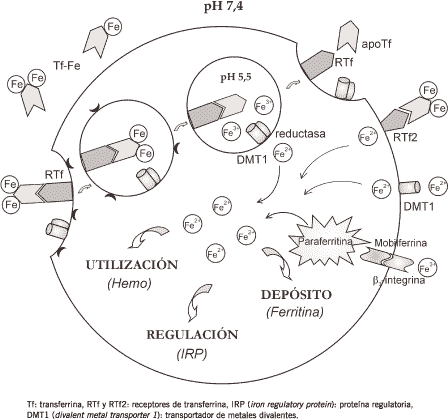
Figura 2. Captación y destino celular de hierro.
Tf: transferrina, RTf y RTf2: receptores de transferrina, IRP (iron regulatory protein): proteína regulatoria, DMT1 (divalent metal transporter 1): transportador de metales divalentes.
EL RECEPTOR DE TRANSFERRINA Y LA HOMEOSTASIS CELULAR DE HIERRO
Los requerimientos de hierro varían según las condiciones celulares particulares en un momento dado. Por ejemplo, las células que se encuentran en una etapa activa de división o aquéllas que tienen requerimientos metabólicos especiales, tales como la producción de hemoglobina, necesitan mayor aporte del metal.
Para mantener la homeostasis de hierro en células de mamíferos es necesario el balance coordinado entre su captación, utilización y almacenamiento intracelular. Ello explica que la expresión de las proteínas clave involucradas en el metabolismo del hierro sea controlada post-transcripcionalmente por los niveles intracelulares del metal. El mecanismo de regulación es mediado por interacciones específicas entre secuencias IRE (iron responsive elements) localizadas en los respectivos ARNm y proteínas citoplasmáticas denominadas IRP (iron regulatory proteins) (25) (Figura 3).
Las IRE, secuencias de nucleótidos filogenéticamente conservadas, están constituidas por 28 bases que forman una estructura secundaria en forma de horquilla (hairpin o stem-loop), conformación que les permite interactuar con las IRP (26).
Estas secuencias IRE están localizadas en las regiones no codificantes o no traducidas (UTRs) situadas en los extremos 5' o 3' de los ARNm y, dependiendo de la posición, difiere el efecto que ocasiona su interacción con las IRP. Las IRE situadas en la región UTR 5' actúan regulando la unión del mensajero al ribosoma o sea, controlando la iniciación de la traducción y aquéllas ubicadas en la región UTR 3', modulan la estabilidad o degradación del ARNm por acción de endorribonucleasas.
En células de mamíferos han sido identificadas dos proteínas IRP (IRP1 e IRP2) que actúan como sensores del contenido celular de hierro.
La IRP1 es una proteína bifuncional que puede actuar como aconitasa citoplasmática o unirse a secuencias IRE. Posee un cluster [4Fe-4S] en su sitio activo y puede convertirse reversiblemente en su forma activa [4Fe-4S] o inactiva [3Fe-4S] en respuesta a modificaciones en la disponibilidad de hierro. Bajo condiciones de depleción del metal, la apoIRP1 puede unirse con alta afinidad a las secuencias IRE. Contrariamente, cuando el aporte de hierro aumenta, la IRP1 incorpora un átomo del metal al cluster [4Fe-4S], adoptando una conformación en la cual es incapaz de interactuar con el ARN, pero posee actividad aconitasa (27). Por lo tanto, las alteraciones en la actividad de la proteína ocurren sin cambios significativos en su concentración.
La IRP2 posee 62% de homología con la IRP1 pero, a diferencia de ella, carece del cluster [4Fe-4S] y no posee actividad aconitasa. Tiene capacidad de unirse con elevada afinidad a secuencias IRE localizadas en los mensajeros. En su región N-terminal contiene una secuencia rica en cisteína que es responsable de la degradación de la proteína vía proteasoma cuando los niveles intracelulares de hierro son altos (28). Por el contrario, cuando el aporte del metal disminuye, se produce la síntesis de novo de la IRP2.
Tal como fue mencionado, la síntesis de las proteínas involucradas en el metabolismo del hierro es regulada a través de la interacción IRE-IRP (Figura 3).
En la región UTR 3' del ARNm del RTf, hay cinco secuencias IRE altamente conservadas. En este caso, la interacción IRE-IRP protege al mensajero de la degradación, permitiendo, de esta manera, su traducción con la consecuente síntesis del receptor (29).
La UTR 5' de los ARNm de las cadenas liviana y pesada de la ferritina (30) y de la enzima d-aminolevulinato sintetasa (ALAS) (31), contienen una única secuencia IRE. En ambos casos, la interacción de las IRP con esta secuencia bloquea la traducción.
De la manera descripta, el sistema IRE-IRP permite a las células regular en forma coordinada la biosíntesis de las proteínas involucradas en la captación (RTf), utilización (ALAS) y almacenamiento (ferritina) de hierro, durante las variaciones fisiológicas de su biodisponibilidad (26). En un estado de depleción de hierro, el objetivo de la célula es incrementar la captación del metal y disminuir su utilización o almacenamiento. Las IRP activas se unen a las secuencias IRE, aumentando la síntesis de RTf mientras que disminuyen la de ferritina y ALAS. Inversamente, cuando los niveles de hierro son altos, el metal es utilizado o almacenado y su incorporación debe ser disminuida. Las IRP se disocian de las IRE y, como consecuencia, el ARNm del RTf es degradado y la síntesis de ferritina y ALAS aumenta.
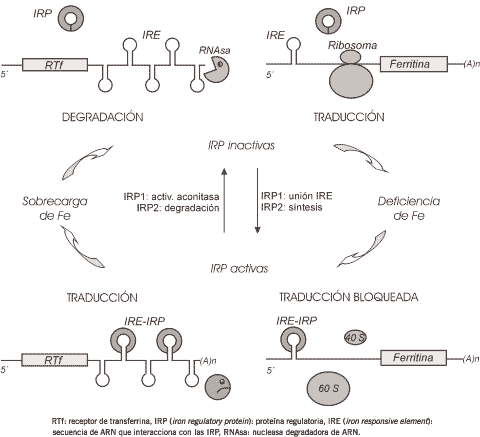
Figura 3. Mecanismo de homeostasis celular de hierro.
RTf: receptor de transferrina, IRP (iron regulatory protein): proteína regulatoria, IRE (iron responsive element): secuencia de ARN que interacciona con las IRP, RNAsa: nucleasa degradadora de ARN.
RECEPTOR DE 2 DE TRANSFERRINA
Existe un segundo receptor de Tf, denominado RTf2, cuya participación en la homeostasis del hierro es menos conocida. La secuencia de aminoácidos de este receptor, deducida a partir de su ARNm, indica que el gen RTf2 codifica para una proteína de membrana que posee 45% de identidad con el RTf en su dominio extracelular (32).
También se han encontrado otras similitudes entre el RTf2 y RTf: ambos receptores pueden unir Tf a nivel de la membrana y mediar la incorporación celular de hierro (32). Además, interactúan con la Tf de una manera dependiente del pH: la holoTf se une a los receptores a pH neutro o levemente alcalino, mientras que la apoTf lo hace sólo a pH ácido. Sin embargo, la afinidad del RTf2 por la holoTf es alrededor de 25 veces menor que la del RTf (33)(34).
La secuencia consenso de internalización del RTf2 no es idéntica a la del RTf. Esto sugiere distintos mecanismos de endocitosis y, probablemente, un procesamiento intracelular diferente (32).
Las regiones no codificantes del ARNm del RTf2 no poseen estructuras similares a las secuencias IRE (32). Esto explicaría que, a diferencia de lo que sucede con el RTf, la expresión del RTf2 no es afectada por los niveles de disponibilidad de hierro. En cambio, ha sido sugerido que podría ser regulada por el ciclo celular (33).
Las diferencias en la distribución tisular de ambos receptores, así como también los distintos perfiles de expresión en células hepáticas y eritroides, sugieren funciones fisiológicas diferentes. Las evidencias disponibles hasta el momento permiten postular que el RTf2 estaría involucrado en el metabolismo del hierro, la función del hepatocito y el proceso de diferenciación eritroide (35)(36).
Según los antecedentes mencionados, las células podrían controlar la captación del metal esencial mediante dos receptores para Tf diferentes: el RTf, de mayor afinidad, cuya expresión es regulada por los niveles celulares de hierro y el RTf2, de baja afinidad, el cual se expresaría dependiendo del ciclo celular o del estatus proliferativo de las células (33).
VÍA DE INCORPORACIÓN DE HIERRO INDEPENDIENTE DE TRANSFERRINA
El mecanismo de incorporación de hierro mediado por Tf ha sido considerado durante mucho tiempo como la única vía de ingreso del metal a las células. Sin embargo, se ha demostrado la existencia de un sistema de captación de hierro independiente de Tf.
Las evidencias más contundentes derivan de trabajos realizados en ratones hipotransferrinémicos. Al nacer, los animales homocigotas para esta mutación, poseen concentraciones de Tf en plasma menores que el 1% de los valores normales y exhiben una anemia microcítica severa. Sin embargo, a pesar de la carencia de Tf, acumulan hierro en tejidos parenquimatosos tales como hígado y páncreas (37).
Como se ha mencionado, prácticamente todo el hierro plasmático circula unido a Tf y el libre es prácticamente indetectable. En desórdenes caracterizados por la sobrecarga de hierro -como por ejemplo la hemocromatosis hereditaria- los sitios de unión de la proteína al metal se encuentran saturados y, bajo estas condiciones, el nivel de hierro no unido a Tf puede alcanzar concentraciones micromolares. Paralelamente, se han encontrado depósitos masivos del metal en células hepáticas.
En las dos patologías mencionadas, ya sea por carencia (hipotransferrinemia) o por saturación (hemocromatosis hereditaria), la disponibilidad de la apoTf es insuficiente para cumplir con la distribución de hierro a las células. Por esta razón, el metal circula en plasma en forma de complejos de bajo peso molecular y es removido mediante un sistema de transporte independiente de Tf (37).
A pesar de que la vía alternativa de captación de hierro ha sido identificada hace más de una década, existe aún discrepancia acerca de cuáles son los mecanismos involucrados, cuál es su regulación, así como también, cuál es su función biológica.
En el entorno celular, el hierro se halla generalmente presente en estado oxidado. Han sido aisladas e identificadas dos proteínas -una de membrana y otra citoplasmática-, involucradas en la captación de iones férricos por la vía independiente de Tf. Las mismas poseen tamaño molecular y características inmunológicas similares a los de las proteínas b3-integrina y mobilferrina. Estas proteínas también han sido caracterizadas como mediadoras de la captación de hierro en células de la mucosa intestinal, las cuales carecen de RTf en su superficie absortiva (38).
Por otra parte, ha sido descripto un mecanismo para la incorporación de iones ferrosos, el cual también podría traslocar el hierro presente en estado férrico, dependiendo de un paso previo de reducción (39-41). La actividad ferri-reductasa sería, por lo tanto, la limitante de la velocidad del proceso (42). Este sistema de transporte es funcionalmente similar al que media la liberación de hierro desde el endosoma al citosol (43). Efectivamente, la proteína transportadora transmembrana ha sido caracterizada e identificada en células eritroides de ratones normales y anémicos como DMT1 (DCT1, Nramp2), la cual, tal como ya fue descripto, media la absorción de iones ferrosos dietarios por parte de los enterocitos (44).
La incorporación de hierro no unido a Tf ha sido caracterizada como un proceso saturable, dependiente del tiempo y de la temperatura de incubación (37)(43). Su actividad no es mediada por endocitosis y no parece ser afectada por cambios en la tasa de crecimiento celular (45).
Varias evidencias demuestran que la regulación del sistema alternativo de transporte de hierro es diferente de la del proceso que involucra al RTf.
En cultivos celulares realizados en presencia de iones férricos o ferrosos se observó un incremento de la actividad de transporte de 59Fe, dependiente del tiempo y de las concentraciones empleadas (42)(45-47). La modulación es muy rápida e independiente de la síntesis de proteínas, lo cual sugiere que se trataría de una regulación a nivel post-traduccional. En ese contexto, se ha postulado que la presencia de hierro extracelular induciría la aparición en membrana de transportadores presentes en un pool críptico (45).
Por otra parte, a diferencia del mecanismo que regula la expresión del RTf, una importante depleción de hierro no desencadena una regulación positiva de la vía independiente de Tf. Esta afirmación se sustenta en ensayos en los cuales la captación del metal por células tratadas con el compuesto quelante de hierro desferrioxamina, no difirió significativamente de la incorporada por células desarrolladas con suplemento normal de hierro (45)(47).
En experiencias realizadas con distintas líneas celulares se ha observado que la captación de hierro independiente de Tf puede ser parcialmente bloqueada por algunos metales de transición di o trivalentes, aunque los resultados son contradictorios. Metales de transición tales como Cu2+, Zn2+, Cd2+ y Mn2+ ejercieron este efecto en células HeLa y K562 (37)(38). Sin embargo, en otros trabajos, los metales trivalentes Al3+, Ga3+ y Cr3+, pero no cationes divalentes, fueron capaces de bloquear la incorporación de hierro no unido a Tf (41)(43). Resultados de nuestro laboratorio demostraron que la vía alternativa de captación de hierro de células K562 no es compartida con el Al3+, a pesar de que este metal no esencial es capaz de ingresar a las células tanto cuando es transportado por Tf como por citrato (47).
Cabe preguntarse en este punto, ¿cuál es la función de la vía alternativa de captación de hierro? En presencia de concentraciones adecuadas de Tf, esta ruta probablemente transporte cantidades mínimas del metal esencial y sirva como un mecanismo para que otros metales ingresen a las células sin competir con el hierro (38)(47). En aquellas situaciones en que la concentración de hierro en plasma excede la capacidad de unión de la Tf circulante, la vía independiente de Tf adquiriría un papel esencial para su remoción del entorno celular.
Ha sido reportado que bajo condiciones de sobrecarga de hierro se produce un aumento de la captación del metal no unido a Tf por parte de los macrófagos. Al considerar que estas células sirven como un sitio de almacenamiento de hierro y cumplen un rol importante en la regulación de la disponibilidad del metal para otras células, este hallazgo ha sido interpretado como una forma de remover del entorno celular las moléculas de hierro potencialmente tóxicas, previniendo por lo tanto posibles daños tisulares (45)(46). Resultados obtenidos por otros autores concuerdan en atribuirle a la vía de captación de hierro independiente de Tf una función primaria en el proceso de detoxificación (38)(42).
En resumen, las células pueden incorporar hierro tanto cuando está unido a Tf (a través del RTf y RTf2) como cuando circula en forma de compuestos de bajo peso molecular (siendo transportado por DMT1 o b3-integrina). Las distintas vías de captación de hierro descriptas en esta revisión se esquematizan en la Figura 2.
DESÓRDENES DEL METABOLISMO DE HIERRO
El contenido total de hierro de un adulto normal es de aproximadamente 3,5 a 5 g. En individuos con un estado nutricional óptimo, alrededor del 65% se encuentra formando parte de la hemoglobina, el 15% está contenido en la mioglobina y diversas enzimas, el 20% está como hierro de depósito y sólo entre 0,1 y 0,2% se encuentra unido a la Tf circulante.
Tal como ya ha sido mencionado, el hierro tiene capacidad de aceptar y donar electrones fácilmente a través de su interconversión entre las formas Fe2+ y Fe3+, propiedad que lo convierte en un compuesto súmamente útil. Sin embargo, este metal también puede provocar daños en los tejidos catalizando la conversión de peróxido de hidrógeno en radicales libres que pueden dañar las membranas celulares, las proteínas o el ADN. Por lo tanto, la homeostasis de hierro se halla estrictamente controlada, ya que tanto la deficiencia como la sobrecarga de este metal esencial son perjudiciales. Sin embargo, el espectro de los desórdenes conocidos del metabolismo del hierro se ha expandido en los últimos años, encontrándose estas enfermedades entre las más comunes de los seres humanos.
Deficiencia de hierro
La deficiencia de hierro se produce cuando los requerimientos del organismo exceden los aportes. En aquellas situaciones en que existe escaso hierro disponible, se producen limitaciones en la síntesis de compuestos fisiológicamente activos que lo contienen y, por lo tanto, surgen consecuencias deletéreas. Por ejemplo, cuando el hierro incorporado resulta insuficiente para la síntesis de hemoglobina, sobreviene anemia de tipo ferropénico (21).
Las causas que pueden conducir a la deficiencia de hierro involucran cuatro factores principales: a) pérdida de sangre (uterina en las mujeres, gastrointestinal en ambos sexos); b) dieta deficiente o inadecuada (pobre en hierro o con exceso de inhibidores de la absorción del metal); c) aumento de la demanda (embarazo) y d) mala absorción (patologías gastrointestinales) (48).
En un principio, las necesidades pueden ser cubiertas recurriendo a las reservas de hierro asociado a ferritina y hemosiderina; pero si el balance negativo persiste, dichas reservas terminan por agotarse.
Como respuesta a una importante disminución del pool intracitoplasmático de hierro, se induce el aumento de la expresión de los RTf a través del mecanismo mediado por la interacción IRE-IRP descripto anteriormente. De esta manera, las células se adecuan para captar el hierro necesario para cubrir sus requerimientos.
Bajo condiciones de deficiencia de hierro, su absorción intestinal también se incrementa como consecuencia de distintas adaptaciones:
• La expresión de la proteína DMT1, mediadora del transporte de iones ferrosos desde el lumen intestinal al interior de los enterocitos, es regulada positivamente (49). El gen que codifica para esta proteína produce dos transcriptos diferentes por splicing alternativo (50). Sólo la isoforma I, que es expresada en la porción proximal del duodeno, contiene una secuencia IRE en la región UTR 3' de su ARNm. Esta secuencia sería responsable del aumento de la expresión de DMT1 como consecuencia de la deprivación de hierro dietario. La expresión de la reductasa férrica duodenal DcytB, la cual es una hemoproteína, aumenta también bajo condiciones de deficiencia de hierro (5). Contrariamente, ha sido reportado que luego de una dosis oral elevada del metal se produce una rápida disminución de la expresión de DMT1 y DcytB (51).
• Las células absortivas de sujetos con deficiencia del metal esencial contienen elevados niveles de hemooxigenasa (11), sugiriendo un aumento del aporte de hierro de origen hémico.
Sobrecarga de hierro
Un estado de sobrecarga en el contenido total de hierro resulta de un aporte del metal que excede los requerimientos. Como las necesidades son limitadas y los seres humanos carecen de un mecanismo fisiológico para la excreción del exceso de hierro, un incremento sostenido en su absorción puede, eventualmente, resultar en una acumulación del metal. Como consecuencia, si la capacidad del organismo para llevar a cabo su transporte y almacenamiento es excedida, la toxicidad del metal puede producir daño a distintos órganos e, inclusive, desencadenar la muerte. Las manifestaciones tóxicas de la sobrecarga de hierro dependen en parte de la magnitud del exceso, de la velocidad de acumulación y de la partición del metal entre sitios de depósito relativamente benignos en los macrófagos y los más perjudiciales en las células parenquimatosas (3)(18)(52).
¿Cómo responden los mecanismos que regulan la homeostasis celular de hierro ante una sobrecarga del metal? Contrariamente a lo que sucede bajo condiciones de deficiencia de hierro, el aumento de la saturación de Tf sérica induce la regulación negativa de la expresión de RTf en células hepáticas y de otros órganos. Por otra parte, el excedente de hierro que circula en la forma de complejos de bajo peso molecular, induciría el aumento de la captación del metal por la vía independiente de Tf en aquellas células encargadas de la detoxificación del medio, como los macrófagos (46).
En respuesta a la sobrecarga de hierro, el hígado secreta hepcidina, una hormona proteica que controla los niveles plasmáticos del metal, regulando la absorción de hierro dietario. La unión de hepcidina a la Fpn induce la internalización y posterior degradación de la proteína transportadora. Como consecuencia, disminuye la exportación de hierro desde el enterocito hacia el plasma (12).
La forma más común de sobrecarga de hierro es la hemocromatosis hereditaria (53). Los pacientes con este desorden autosómico recesivo absorben regularmente dos o tres veces más hierro dietario que las personas normales. Por lo tanto, la cantidad del metal unido a Tf aumenta, pudiendo alcanzar valores de 50-60% de saturación. El exceso de hierro es depositado en las células parenquimatosas del hígado, corazón, páncreas, glándula pituitaria y paratiroides. Como consecuencia, los pacientes desarrollan hepatomegalia con fibrosis y cirrosis, cardiomiopatías y diversas endocrinopatías como diabetes mellitus, hipopituitarismo, hipogonadismo e hipoparatiroidismo (54).
Curiosamente, esta enfermedad no es causada por un defecto en una proteína involucrada en el transporte de hierro, sino por una anomalía en un regulador del transporte. En 1996 fue identificada la proteína de membrana HFE que se halla mutada en la mayoría de los pacientes con hemocromatosis hereditaria (55). Esta proteína, estructuralmente similar a las del complejo mayor de histocompatibilidad de clase I, no une ni transporta hierro. Parece ser una molécula regulatoria que influye sobre la eficiencia de la absorción del metal esencial.
HFE forma un heterodímero con la b2-microglobulina, el cual es expresado en la superficie de muchas células, incluyendo las de la cripta duodenal y macrófagos. A pH neutro, el heterodímero forma un complejo estable con el RTf (56), resultando en un cambio conformacional del receptor que provoca modificación de su afinidad por Tf (57). De esta manera, HFE modularía la captación de hierro unido a Tf desde el plasma hacia los enterocitos de la cripta duodenal y participaría en el mecanismo por el cual dichas células detectan modificaciones de las reservas corporales de hierro (58).
La alteración de este mecanismo desempeña un rol fundamental en la patogénesis de la hemocromatosis hereditaria. Han sido descriptas mutaciones en el gen HFE que atenúan la captación de hierro unido a Tf por parte de las células duodenales de la cripta, proveyendo de esta manera señales paradójicas que conducen a la regulación positiva de la expresión de DMT1 en las células absortivas, a pesar de la elevada saturación de la Tf y del alto contenido hepático de hierro (59)(60).
Hierro y anemia de la enfermedad crónica
La anemia de la enfermedad crónica es una condición adquirida que afecta a pacientes con una variedad de desórdenes inflamatorios como infecciones, artritis, neoplasias, etc. La patogénesis de esta anemia ha sido atribuida a deficiencias en varias etapas de la eritropoyesis: inhibición de la respuesta de los progenitores eritroides a la eritropoyetina, disminución de la vida media de los eritrocitos maduros y reducción de la disponibilidad de hierro (61)(62).
A pesar de hallarse adecuadas o aun incrementadas las reservas de hierro, bajos valores de ferremia caracterizan esta enfermedad. Estas manifestaciones parecen derivar de una disminuida absorción intestinal de hierro, así como de la retención del metal en los macrófagos reticuloendoteliales. Aunque la anemia aumenta la morbilidad de la enfermedad primaria, probablemente forma parte de un mecanismo de defensa del huésped destinado a secuestrar hierro y luchar de esta manera contra las infecciones y el cáncer. Sin embargo, también conduce a la deficiencia en la disponibilidad del metal para la eritropoyesis, ya que no puede ser eficientemente movilizado desde los lugares de depósito hacia los precursores eritroides (63).
Aunque las bases moleculares de la anemia de la enfermedad crónica no han sido totalmente dilucidadas aún, determinadas citoquinas proinflamatorias y proteínas de fase aguda parecen estar implicadas en su desarrollo (62)(64)(65).
En los últimos años el péptido hepcidina ha sido considerado un mediador clave de la patogénesis de la anemia de la enfermedad crónica (62-67). Tal como ha sido descripto anteriormente (ver "Vías de absorción intestinal de hierro"), los hepatocitos regulan el metabolismo del hierro en forma endocrina a través de la secreción de esta hormona proteica.
La proteína Fpn, la cual actúa como el receptor de hepcidina, es expresada en varios tipos celulares que exportan hierro al plasma (enterocitos, células placentarias, hepatocitos y macrófagos) (12). Por lo tanto, además de regular la absorción intestinal del hierro dietario, la hepcidina controla la transferencia materno-fetal del metal durante el embarazo, el movimiento del hierro almacenado en los hepatocitos y la liberación del catión reciclado en los macrófagos.
Bajo condiciones de anemia o hipoxia, cesa la producción hepática de hepcidina, aumentando por lo tanto el transporte basolateral de hierro y la movilización desde los lugares de depósito a través de la Fpn. Inversamente, cuando el porcentaje de saturación de la Tf es elevado, se induce la síntesis de hepcidina. Como cosecuencia, se produce la internalización y degradación de la Fpn, disminuyendo de esta manera la liberación de hierro desde los enterocitos y macrófagos (14).
Además de responder a los niveles de hierro, se ha observado que la secreción de hepcidina es inducida durante los procesos inflamatorios. En presencia de interleuquina 6 (citoquina proinflamatoria) los hepatocitos liberan hepcidina como parte de la respuesta de fase aguda de tipo II (64). El efecto producido por esta hormona proteica sobre el transporte de hierro causa la característica disminución de la concentración plasmática del metal: hipoferremia de la inflamación. Aunque el objetivo primario de esta respuesta sería aumentar la resistencia del huésped a la infección mediante la restricción de la disponibilidad de hierro, contribuye también a la anemia de la enfermedad crónica.
La hepcidina constituye por lo tanto el vínculo molecular entre la regulación del metabolismo del hierro, los procesos inflamatorios y la anemia.
PERSPECTIVAS
A pesar de la profusa investigación de los mecanismos involucrados en la absorción y captación celular de hierro surgen todavía numerosos interrogantes, en particular, los relacionados con los procesos de regulación: ¿cuál es el significado fisiológico de las vías de captación de hierro independientes de Tf ?, se relaciona la regulación y/o respuesta de esta vía con señales específicas dependientes de la disponibilidad de hierro?, ¿cuál es la influencia de procesos que involucran estrés oxidativo e inflamación sobre la homeostasis del hierro?, ¿cuáles son las señales de activación y cómo funcionan las distintas proteínas que parecen regular los niveles celulares de hierro?, ¿cuáles son las bases moleculares del metabolismo del hierro en células de tejidos especializados, tales como el eritropoyético y el nervioso? Las respuestas a estos interrogantes contribuirán a ampliar el conocimiento de los mecanismos que regulan la homeostasis celular del hierro.
Por otra parte, si la hepcidina constituye un factor central en el metabolismo del hierro, la modulación de su bioactividad podría constituir una nueva estrategia para el tratamiento de pacientes con desórdenes de dicho metabolismo. La utilización de hepcidina, de sus agonistas o de sustancias que estimulen su producción por parte del hígado puede ser propuesta como un posible tratamiento de las enfermedades de sobrecarga de hierro. Contrariamente, los agonistas de la hepcidina podrían ser utilizados en el tratamiento de la anemia de la enfermedad crónica.
Agradecimientos
Este trabajo de actualización fue realizado en el marco de las investigaciones del laboratorio financiadas por la Universidad de Buenos Aires, el Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) y la Agencia Nacional de Promoción Científica y Tecnológica (ANPCYT).
Correspondencia
DRA. GLADYS PÉREZ
Departamento de Química Biológica
Facultad de Ciencias Exactas y Naturales, UBA
Pabellón II, Piso 4, Ciudad Universitaria
(1428) CIUDAD AUTÓNOMA DE BUENOS AIRES
Fax: 011-4576-3342
E-mail: gperez@qb.fcen.uba.ar
Referencias bibliográficas
1. Aisen P, Listowsky I. Iron and storage proteins. Annu Rev Biochem 1980; 49: 357-93.
2. Ponka P. Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 1997; 89: 1-25.
3. Crichton RR, Wilmet S, Legssyer R, Ward RJ. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J Inorg Biochem 2002; 91: 9-18.
4. Conrad ME, Umbreit JN, Peterson RD, Moore EG, Harper KP. Function of integrin in duodenal mucosal uptake of iron. Blood 1993; 81: 517-21.
5. Latunde-Dada GO, Van der Westhuizen J, Vulpe CD, Anderson GJ, Simpson RJ, McKie AT. Molecular and functional roles of duodenal cytochrome B (Dcytb) in iron metabolism. Blood Cells Mol Dis 2002; 29: 356-60.
6. Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci USA 1998; 95: 1148-53.
7. Uzel C, Conrad ME. Absorption of heme iron. Semin Hematol 1998; 35: 27-34.
8. Worthington MT, Cohn SM, Miller SK, Luo RQ, Berg CL. Characterization of a human plasma membrane heme transporter in intestinal and hepatocyte cell lines. Am J Physiol Gastrointest Liver Physiol 2001; 280: 1172-7.
9. Umbreit JN, Conrad ME, Moore EG, Desai MP, Turrens J. Paraferritin: a protein complex with ferrireductase activity is associated with iron absorption in rats. Biochemistry 1996; 35: 6460-9.
10. Kaplan J. Mechanisms of cellular iron acquisition: another iron in the fire. Cell 2002; 111: 603-6.
11. Conrad ME, Umbreit JN. Iron absorption and transport-an update. Am J Hematol 2000; 64: 287-98.
12. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, McVey Ward D, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306: 2090-3.
13. Leong W-I, Lönerdal B. Hepcidin, the recently identified peptide that appears to regulate iron absorption. J Nutr 2004; 134: 1-4.
14. Muñoz Gómez M, Campos Garríguez A, García Erce JA, Ramírez Ramírez G. Fisiopatología del metabolismo del hierro: implicaciones diagnósticas y terapéuticas. Nefrología 2005; 25: 9-19.
15. Baker EN, Lindley PF. New perspectives on the structure and function of transferrins. J Inorg Biochem 1992; 47: 147-60.
16. Pakdaman R, El Hage Chahine JM. A mechanism for iron uptake by transferrin. Eur J Biochem 1996; 236: 922-31.
17. Bali PK, Aisen P. Receptor-induced switch in site-site cooperativity during iron release by transferrin. Biochemistry 1992; 31: 3963-7.
18. Van Eijk HG, De Jong G. The physiology of iron, transferrin, and ferritin. Biol Trace Element Res 1992; 35: 13-24.
19. Huebers HA, Finch CA. The physiology of transferrin and transferrin receptors. Physiol Rev 1987; 67: 520-82.
20. Beguin Y. The soluble transferrin receptor: biological aspects and clinical usefulness as quantitative measure of erythropoiesis. Haematologica 1992; 77: 1-10.
21. Brittenham GM. Disorders of Iron Metabolism: Iron deficiency and overload. En Hoffman R, Benz EJ, Shattil SJ, Furie B, Cohen HJ, Silberstein LE, McGlave P, editors. Hematology: Basic Principles and Practice. 3rd ed. New York: Churchill Livingstone, 2000; p. 397-427.
22. Wileman T, Harding C, Stahl P. Receptor-mediated endocytosis. Biochem J 1985; 232: 1-14.
23. Gruenheid S, Canonne-Hergaux F, Gauthier S, Hackam DJ, Grinstein S, Gros P. The iron transport protein NRAMP2 is an integrated membrane glycoprotein that colocalizes with transferrin in recycling endosomes. J Exp Med 1999; 189: 831-41.
24. Young SP, Bomford A. Iterative endocytosis of transferrin by K562 cells. Biochem J 1994; 298: 165-70.
25. Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 1993; 72: 19-28.
26. Hentze MW, Kühn LC. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci, USA 1996; 93: 8175-82.
27. Kaldy P, Menotti E, Moret R, Kühn LC. Identification of RNA-binding surfaces in iron regulatory protein-1. EMBO J 1999; 18: 6073-83.
28. Iwai K, Klausner RD, Rouault TA. Requeriments for iron-regulated degradation of the RNA binding protein, iron regulatory protein 2. EMBO J 1995; 14: 5350-7.
29. Casey JL, Hentze MW, Koeller DM, Caughman SW, Rouault TA, Klausner RD, et al. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science 1988; 240: 924-8.
30. Leibold EA, Munro HN. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5' untranslated region of ferritin heavy- and light-subunit mRNAs. Proc Natl Acad Sci USA 1988; 85: 2171-5.
31. Cox TC, Bawden MJ, Martin A, May BK. Human erythroid 5-aminolevulinate synthase: promoter analysis and identification of an iron-responsive element in the mRNA. EMBO J 1991; 10: 1891-902.
32. Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. J Biol Chem 1999; 274: 20826-32.
33. Kawabata H, Germain RS, Vuong PT, Nakamaki T, Said JW, Koeffler HP. Transferrin receptor 2-a supports cell growth both in iron-chelated cultured cells and in vivo. J Biol Chem 2000; 275: 16618-25.
34. West AP, Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem 2000; 275: 38135-8.
35. Fleming RE, Migas MC, Holden CC, Waheed A, Britton RS, Tomatsu S, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci USA 2000; 97: 2214-9.
36. Kawabata H, Germain RS, Ikezoe T, Tong X, Green EM, Gombart AF, et al. Regulation of expression of murine transferrin receptor 2. Blood 2001; 98: 1949-54.
37. Sturrock A, Alexander J, Lamb J, Craven CM, Kaplan J. Characterization of a transferrin-independent uptake system for iron in HeLa cells. J Biol Chem 1990; 265: 3139-45.
38. Conrad ME, Umbreit JN, Moore EG, Uzel C, Berry MR. Alternate iron transport pathway. J Biol Chem 1994; 269: 7169-73.
39. Jordan I, Kaplan J. The mammalian transferrin-independent iron transport system may involve a surface ferrireductase activity. Biochem J 1994; 302: 875-9.
40. Akompong T, Inman RS, Wessling-Resnick M. Phorbol esters stimulate non-transferrin iron uptake by K562 cells. J Biol Chem 1995; 270: 20937-41.
41. Attieh ZK, Mukhopadhyay CK, Seshadri V, Tripoulas NA, Fox PL. Ceruloplasmin ferroxidase activity stimulates cellular iron uptake by a trivalent cation-specific transport mechanism. J Biol Chem 1999; 274: 1116-23.
42. Randell EW, Parkes JG, Olivieri NF, Templeton DM. Uptake of non-transferrin-bound iron by both reductive and nonreductive processes is modulated by intracellular iron. J Biol Chem 1994; 269: 16046-53.
43. Inman RS, Wessling-Resnick M. Characterization of transferrin-independent iron transport in K562 cells. J Biol Chem 1993; 268: 8521-8.
44. Canonne-Hergaux F, Zhang AS, Ponka P, Gros P. Characterization of the iron transporter DMT1 (NRAMP 2/DCT1) in red blood cells of normal and anemic mk/mk mice. Blood 2001; 98: 3823-30.
45. Kaplan J, Jordan I, Sturrock A. Regulation of the transferrin-independent iron transport system in cultured cells. J Biol Chem 1991; 266: 2997-3004.
46. Olakanmi O, Stokes JB, Pathan S, Britigan BE. Polyvalent cationic metals induce the rate of transferrin-independent iron acquisition by HL-60 cells. J Biol Chem 1997; 272: 2599-606.
47. Pérez G, Pregi N, Vittori D, Di Risio C, Garbossa G, Nesse A. Aluminum exposure affects transferrin-dependent and -independent iron uptake by K562 cells. Biochim Biophys Acta 2005; 1745: 124-30.
48. Chanarin I. Anaemia and red blood cell hyperplasia. En: McGee J, Isaacson P, Wright N, editors. Oxford Textbook of Pathology. New York: Oxford University Press; 1992. p. 1692-5.
49. Canonne-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 1999; 93: 4406-17.
50. Lee PL, Gelbart T, West C, Halloran C, Beutler E. The human Nramp2 gene: characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol Dis 1998; 24: 199-215.
51. Frazer DM, Wilkins SJ, Becker EM, Murphy TL, Vulpe CD, McKie AT, et al. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 2003; 52: 340-6.
52. Chapman RW. Iron overload of the liver. En: McGee J, Isaacson P, Wright N, editors. Oxford Textbook of Pathology. New York: Oxford University Press; 1992. p. 1369-72.
53. Roy CN, Andrews NC. Recent advances in disorders of iron metabolism: mutations, mechanisms and modifiers. Hum Mol Genet 2001; 10: 2181-6.
54. Andrews NC. Disorders of iron metabolism. N Engl J Med 1999; 341: 1986-95.
55. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13: 399-408.
56. Waheed A, Parkkila S, Saarnio J, Fleming RE, Zhou XY, Tomatsu S, et al. Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum. Proc Natl Acad Sci USA 1999; 96:1579-84.
57. Roy CN, Penny DM, Feder JN, Enns CA. The hereditary hemochromatosis protein, HFE, specifically regulates transferrin-mediated iron uptake in HeLa cells. J Biol Chem 1999; 274: 9022-8.
58. Waheed A, Grubb JH, Zhou XY, Tomatsu S, Fleming RE, Costaldi ME, et al. Regulation of transferrin-mediated iron uptake by HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci USA 2002; 99: 3117-22.
59. Fleming RE, Migas MC, Zhou X, Jiang J, Britton RS, Brunt EM, et al. Mechanism of increased iron absorption in murine model of hereditary hemochromatosis: increased duodenal expression of the iron transporter DMT1. Proc Natl Acad Sci USA 1999; 96: 3143-8.
60. Stuart KA, Anderson GJ, Frazer DM, Powell LW, McCullen M, Fletcher LM, et al. Duodenal expression of iron transport molecules in untreated haemochromatosis subjects. Gut 2003; 52: 953-9.
61. Weiss G. Pathogenesis and treatment of anaemia of chronic disease. Blood Reviews 2002; 16: 87-96.
62. Means RT. Recent developments in the anemia of chronic disease. Curr Hematol Rep 2003; 2: 116-21.
63. Roy CN, Weinstein DA, Andrews NC. 2002 E. Mead Johnson Award for Research in Pediatrics Lecture: the Molecular Biology of the Anemia of Chronic Disease: A Hypothesis. Pediatr Res 2003; 53: 507-12.
64. Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101: 2461-3.
65. Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood 2002; 100: 3776-81.
66. Del Castillo Rueda A, De Portugal Alvarez J. Hepcidina, una nueva proteína en la homeostasis del hierro. An Med Intern (Madrid) 2003; 20: 605-6.
67. Roy CN, Andrews NC. Anemia of inflammation: the hepcidin link. Curr Opin Hematol 2005; 12: 107-11.
Aceptado para su publicación el 9 de agosto de 2005

