










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. v.41 n.3 La Plata jul./sep. 2007
BIOQUÍMICA CLÍNICA
Porfiria Congénita Eritropoyética en la Argentina: 4 niños y un caso de manifestación tardía*
Congenital Erythropoietic Porphyria in Argentina: 4 children and one late onset case
María Victoria Rossetti1*, Victoria Estela Parera1*, Viviana Alicia Melito2**, Alcira Batlle1***
1. Dr. en Ciencias Químicas.
2. Lic. en Ciencias Químicas.
* Investigador Independiente de la Carrera del Investigador Científico del CONICET.
** Miembro Principal de la Carrera de Apoyo a la Investigación y Desarrollo del CONICET.
*** Investigador Superior de la Carrera del Investigador Científico del CONICET.
* Centro de Investigaciones sobre Porfirinas y Porfirias - CIPYP, Consejo Nacional de Investigaciones Científicas y Técnicas. Universidad de Buenos Aires. Buenos Aires. Argentina.
Resumen
Las porfirias son la consecuencia de fallas en el metabolismo del hemo. La Porfiria Congénita Eritropoyética (PCE) (enfermedad de Günther) es una porfiria cutánea rara que se transmite en forma autosómica recesiva. Se produce debido a la presencia de mutaciones en el gen de la uroporfirinógeno III sintetasa (UROIII-S) que llevan a una marcada disminución de su actividad y a la producción y acumulación de elevadas cantidades de porfirinas de la serie isomérica I en plasma, tejidos y huesos, responsables de la severa sintomatología cutánea que generalmente presentan los pacientes con esta porfiria. Se han descripto sólo alrededor de 200 casos a nivel mundial. Su expresión clínica es muy heterogénea, encontrándose desde casos muy graves con severo compromiso cutáneo, transfusión-dependiente, hasta casos leves con escasa sintomatología cutánea. Se presentan 5 casos de pacientes argentinos con PCE, 4 infantiles y uno de manifestación tardía, diagnosticados en el Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP), que constituyen, hasta el momento, los únicos registrados en Argentina. Se encontraron elevadas cantidades de porfirinas en plasma, sangre, orina y materia fecal y un patrón de porfirinas con predominio de la serie I. La actividad de la UROIII-S estaba reducida en un 25-44% con respecto al valor normal. El diagnóstico certero y precoz de esta porfiria es fundamental para aplicar tempranamente el tratamiento adecuado en cada caso y brindarle al paciente una mejor calidad de vida.
Palabras clave: Hemo; Porfirinas; Porfiria; Porfiria Congénita Eritropoyética; Uroporfirinógeno III sintetasa
Summary
Porphyrias are metabolism disorders caused by a partial deficiency in one of the heme biosynthetic pathway enzymes. Congenital Erythropoietic Porphyria, also termed Günther disease, is extremely rare and is inherited as an autosomal recessive trait that results from the markedly deficient activity of the fourth enzyme in the heme biosynthetic pathway, Uroporphyrinogen III synthase (UROIII-S). This enzyme deficiency leads to an increased production and accumulation of the nonphysiological and phototoxic type I porphyrins responsible for the typical clinical manifestations. The disease severity is markedly heterogeneous, ranging from severe transfusion dependency throughout life to milder adult cases with only cutaneous photosensitivity. Only 200 cases have been described all over the world so far. In this work five Argentinean CEP patients are presented, 4 infantile and one late onset case, diagnosed in the CIPYP which are, as far as it is known, the only cases described in Argentina. Increased amounts of porphyrins were found in plasma, blood, urine and faeces, together with high amounts of the pathogenic type I isomer. Enzyme activity was reduced to 25-44% respect to normal values. Early diagnosis is important for correct treatment so as to prevent the characteristic mutilation of the disease and to improve patient´s life quality.
Key words: Heme; Porphyrins; Porphyrias; Congenital Erythropoietic Porphyria; Uroporphyrinogen III synthase
INTRODUCCIÓN
Se denominan porfirias al grupo de enfermedades metabólicas causadas por fallas en el metabolismo del hemo. Estos defectos enzimáticos, parciales y primarios, traen como consecuencia sobreproducción y acumulación del sustrato de la enzima afectada, dando lugar a la aparición de diversos síntomas clínicos característicos para cada tipo de porfiria. Se clasifican en hepáticas o eritropoyéticas, según el órgano principal de acumulación de intermediarios del camino del hemo y en agudas y/o cutáneas según la sintomatología clínica asociada (1).
La Porfiria Congénita Eritropoyética (PCE) o enfermedad de Günther, es una porfiria cutánea rara, de carácter autosómico recesivo, provocada por una marcada deficiencia en la actividad de la uroporfirinógeno III sintetasa (Uro III-S) (2-5).
Esta enzima cataliza la ciclación e isomerización del tetrapirrol lineal, hidroximetilbilano (HMB) o preuroporfirinógeno formado por la hidroximetilbilano sintetasa (HMB-S) o porfobilinógeno deaminasa (PBG-D) en la tercera etapa de la biosíntesis del hemo, a uroporfirinógeno III (Urogen III) (6-8). El HMB se cicla en forma muy rápida y no enzimáticamente al tetrapirrol uroporfirinógeno I (Urogen I) (6), por lo tanto, ante una deficiencia de la enzima URO III-S se produce una rápida acumulación de este isómero no fisiológico que es decarboxilado para dar el coproporfirinógeno I (Coprogen I). Ambos compuestos son oxidados no enzimáticamente a sus correspondientes porfirinas, uroporfirina I (URO I) y coproporfirina I (COPRO I), cuya acumulación es altamente tóxica para el organismo. Así, las porfirinas de la serie I, que no llegan al producto final de la ruta biosintética, se acumulan en glóbulos rojos produciendo hemólisis y su liberación al torrente sanguíneo. Las porfirinas liberadas circulan y se depositan en diferentes tejidos y huesos produciendo eritrodoncia y aumentando la fragilidad ósea. Debido a su acción fotooxidativa, las porfirinas circulantes causan gran fotosensibilidad y severos daños en la piel desde la niñez. Entre las múltiples lesiones que pueden encontrarse se observa la presencia de eritema, prurito, hiperpigmentación, hipertricosis, pigmentación de la uñas, alopecía y ampollas en zonas expuestas que se ulceran dando lugar a la aparición de costras que progresan dejando lesiones mutilantes (9). También puede verse comprometida la zona ocular con graves lesiones que pueden derivar en ceguera. La anemia hemolítica, que puede ser severa, se asocia a esplenomegalia y fotofobia. Las porfirinas acumuladas en hueso, hígado, bazo, intestino y riñones se excretan en grandes cantidades por orina y materia fecal (1).
El gen que codifica para la enzima deficiente, URO III-S, está localizado en el cromosoma 10 (10q25.2-q26.1) (10) y hasta el momento se han descripto alrededor de 40 mutaciones diferentes en pacientes con PCE (Human Gene Mutation Database, 2007). La expresión de la mayoría de estas mutaciones en un sistema bacteriano indicó la existencia de una relación genotipo-fenotipo, así la presencia y severidad de las manifestaciones clínicas de la porfiria están relacionadas con la actividad residual de la enzima mutada pudiéndose encontrar desde casos muy severos hasta muy leves de manifestación tardía con sólo compromiso cutáneo y hemólisis compensada (4)(5)(11)(12).
En general, el pronóstico de la enfermedad es malo con una expectativa de vida de no más de 3 ó 4 décadas. Los tratamientos consisten principalmente en evitar la exposición solar y emplear fotoprotección adecuada a fin de minimizar las lesiones cutáneas, transfusiones sanguíneas para reducir la eritropoyesis y esplenectomía para reducir la hemólisis y la necesidad de transfusiones. Es fundamental la protección de la piel de la luz y de los traumatismos y el tratamiento de las infecciones o ulceraciones con antibióticos es esencial para minimizar las mutilaciones (13-15). Para los pacientes transfusión-dependientes las transfusiones crónicas de sangre son fundamentales y se han empleado terapéuticamente en pacientes moderadamente afectados con anemia hemolítica leve a fin de suprimir la eritropoyesis y así disminuir la producción de porfirinas (16). Esta estrategia es efectiva hasta la pubertad, momento en el cual la producción de porfirinas puede aumentar y entonces se hace necesario aplicar un tratamiento más agresivo, con hidroxiurea por ejemplo, a fin de reducir la producción de porfirinas en la médula ósea (17). Sin embargo, estos tratamientos no son generalmente exitosos y en la actualidad el transplante de médula ósea compatible en pacientes severamente afectados, constituye la única opción efectiva al sustituir los eritroblastos de la médula corrigiendo así el defecto enzimático con la reducción de los niveles de porfirinas y consecuente disminución de la sintomatología cutánea (12)(18-24).
Se encuentran descriptos en la literatura internacional menos de 200 casos de PCE de los cuales 13 corresponden a casos de manifestación tardía (25).
Se presenta una recopilación de los casos de Porfiria Congénita Eritropoyética que concurrieron al CIPYP a partir del año 1984 en el cual se diagnosticó el primer caso de PCE en este país. Estos resultados fueron parcialmente publicados previamente (26)(27).
MATERIALES Y MÉTODOS
Para el diagnóstico de la porfiria se realizaron las determinaciones en muestras de orina de 24 horas, materia fecal, glóbulos rojos, sangre y plasma que se indican a continuación, según la metodología descripta por Batlle (28). Las porfirinas plasmáticas (IPP) se determinaron fluorométricamente y las porfirinas en orina (PTO), en eritrocitos (PTS) y en materia fecal (PTMF) se midieron espectrofotométricamente luego de su extracción por cromatografía de intercambio iónico en orina y por solventes orgánicos en los otros dos casos. El patrón de excreción de porfirinas se obtuvo por cromatografía en capa delgada y/o cromatografía líquida de alta presión (HPLC). Los precursores urinarios se cuantificaron por espectrofotometría luego de su extracción por cromatografía de intercambio iónico (28).
La determinación de la actividad de URO III-S eritrocitaria se realizó mediante la técnica de ensayo acoplado (29). El análisis del tipo isomérico se realizó por HPLC mediante adaptación de la técnica de Lim y col (30) o según la metodología de Falk y Benson (31).
La determinación de la actividad enzimática de la URO-D eritrocitaria se llevó a cabo según la metodología descripta por Afonso y col. (32).
CASOS CLíNICOS
Todos los pacientes estudiados concurrieron o enviaron sus muestras al CIPYP derivados de diferentes Centros de Salud del país con diagnóstico presuntivo de porfiria y fueron interrogados acerca del comienzo de la sintomatología, historia familiar de la enfermedad, otras patologías y eventuales tratamientos recibidos. De cada paciente se obtuvo consentimiento escrito para su estudio.
Los pacientes PCE-1 y PCE-2 eran hermanos provenientes de la ciudad de Tartagal (Salta) y concurrieron por primera vez a la consulta a la edad de 3 y 2 años respectivamente, en 1984. A los 15 meses PCE-1 comenzó con eritrodoncia luego de haber recibido antibióticos por una diarrea. A los 18 meses los padres refieren la aparición de pañales oscuros. A los 2 años, en la era estival, aparecieron las primeras ampollas con contenido seroso en cara, dorso de manos, pies y otras áreas expuestas e hipertricosis siendo tratada como una alergia.
En octubre de 1984, en una nueva consulta dermatológica, se piensa en un caso de porfiria indicándose el uso de pantalla solar. En el mes de noviembre PCE-1 fue internada en el Hospital de Niños de Salta enviándose las primeras muestras al CIPYP.
A los tres años, la paciente presentaba hipertricosis malar y frontal, lesiones erosivo-costrosas en región malar derecha, eritrodoncia, lesiones eritematosas residuales en tórax anterior. También presentaba hipertricosis, múltiples lesiones eritematosas pequeñas y secuelas de ampollas en brazos y desde la rodilla hacia el dorso del pie máculas pigmentarias y eritematosas. Debido a su esplenomegalia fue sometida a una esplenectomía con el objetivo, entre otros, de reducir la intensa hemólisis, lográndose una mejoría transitoria.
El paciente PCE-2 presentó eritrodoncia al año de edad y a partir del año y medio aparecieron lesiones eritematovesiculosas muy pruriginosas en la cara, orinas rojas, hipertricosis malar y en la frente, múltiples lesiones erosivo-costrosas y máculas pigmentarias residuales en zonas malares. También se observaron lesiones y ampollas en manos y esplenomegalia.
La paciente PCE-3 concurrió al CIPYP por primera vez en 1994 a los 2 meses de vida sufriendo, desde el nacimiento, hemorragias generalizadas, por lo que se la sometió a reiteradas transfusiones con sangre entera y plasma, administrándole además vitamina K, colesteramina y antibióticos. A la edad de un mes presentaba orinas oscuras y hemorragia fecal y urinaria, hígado y bazo de tamaño aumentado con ecoestructura no homogénea no refringente. Vesícula, páncreas y riñones sin alteraciones. El análisis de sangre reveló un hematocrito de 15% con alto porcentaje (1,9%) de reticulocitos, concordante con una importante anemia hemolítica. Presentaba, además, edema transitorio de miembros inferiores y párpados.
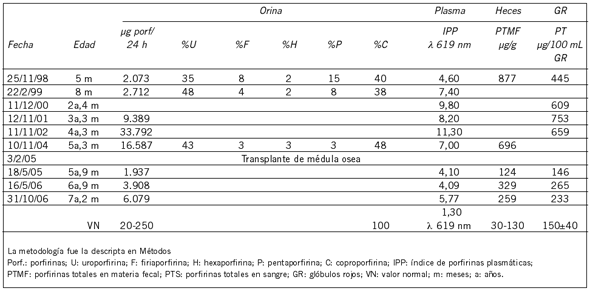
Las muestras del paciente PCE-4 fueron derivadas al CIPYP por primera vez en noviembre de 1998 cuando contaba con sólo 5 meses de edad, presentando manifestaciones cutáneas leves con ampollas con contenido seroso y millia desde el nacimiento. Inicialmente se lo trató con pantallas solares. A los 6 años, cuando la manifestación cutánea era más severa fue sometido a un transplante de médula ósea, utilizando como donante compatible a su hermana mayor.
El paciente adulto de 57 años (PCE-5) acudió por primera vez al CIPYP en 2001 con diagnóstico presuntivo de PCT presentando orinas oscuras y sintomatología cutánea, de 2 años de evolución, consistente en ampollas, hipertricosis y leve fragilidad. Mostraba, además, cicatrices estrelladas con reborde hiperpigmentado en ambas piernas y lesiones costrosas con secreción acuosa en cuero cabelludo. No se observó visceromegalia. Se le recomendó como tratamiento el uso de pantallas solares, dieta rica en hidratos de carbono y cloroquina (Nivaquine). Al año se inició la terapia combinada consistente en S-adenosil-L-metionina (Transmetil) y cloroquina. El paciente padece también de talasemia menor por lo que recibe ácido fólico.
RESULTADOS
Los pacientes infantiles PCE-1, PCE-2, PCE-3 y PCE-4 presentaron síntomas de porfiria desde su nacimiento, con excreción muy elevada de porfirinas en orina y materia fecal. El contenido de porfirinas en glóbulos rojos y plasma también fue muy alto (Tabla I) (Tabla II) (Tabla III).
Tabla I. Datos bioquímicos de los pacientes PCE-1 y PCE-2.

Tabla II. Evolución de la paciente PCE-3.
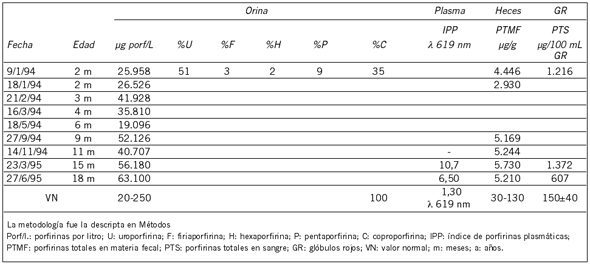
Tabla III. Evolución del paciente PCE-4.
En el caso de los pacientes PCE-1 y PCE-2 la identificación del tipo isómerico se llevó a cabo mediante la técnica de cromatografía en papel (31) observándose una excreción masiva de uro y coproporfirinas por orina con claro predominio de la serie I, típico perfil correspondiente a una PCE (Tabla I). La paciente PCE-1 fue sometida a una esplenectomía y el estudio de una biopsia del órgano indicó la presencia de un contenido elevado de porfirinas nuevamente con predominio de la serie I, corroborando el diagnóstico de PCE (Tabla I). Actualmente se sabe que ambos hermanos se encuentran vivos pero sin efectuarse controles bioquímicos desde hace 20 años.
La paciente PCE-3 presentó desde su nacimiento niveles muy elevados de porfirinas en orina, materia fecal y plasma los cuales permanecieron sin cambios hasta su deceso ocurrido a los 22 meses de edad. En esta paciente la determinación del tipo isomérico se llevó a cabo por HPLC, de acuerdo a lo descripto en Materiales y Métodos. Los resultados obtenidos se muestran en la Figura 1, observándose un elevado porcentaje de la serie no fisiológica.
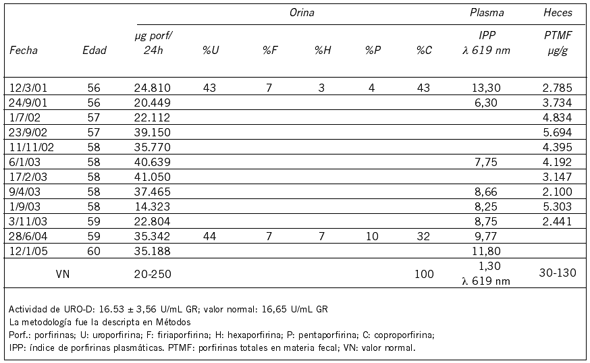
El paciente PCE-4 también fue diagnosticado en sus primeros meses de vida con un cuadro compatible con PCE y sintomatología cutánea moderada. Su diagnóstico se realizó mediante la evaluación del contenido y patrón de porfirinas presentes en orina, heces, sangre y plasma (Tabla III). El tipo isomérico de las porfirinas urinarias determinado por HPLC indicó una proporción de 96-98% de URO I a 2-4% de URO III (Fig. 2). La Tabla III muestra el seguimiento de los parámetros bioquímicos pre y post transplante de médula ósea, en el que se observa una importante disminución de los mismos, sin normalización, a los 3 meses de efectuado el mismo. Sin embargo, los estudios bioquímicos realizados a los 15 y 27 meses post transplante indicaron un aumento del contenido de porfirinas principalmente en orina y plasma. A pesar de ello, y de no estar recibiendo ninguna medicación, el paciente continúa con una buena evolución clínica y sin lesiones cutáneas.
En el caso del paciente adulto (PCE-5) los estudios bioquímicos realizados (Tabla IV) (Fig. 2), permitieron realizar el diagnóstico diferencial de PCE ya que el paciente había sido derivado al CIPYP como un posible caso de PCT. El análisis de las porfirinas urinarias por HPLC reveló un patrón claramente compatible con una PCE (96-98% URO I, 2-4% URO III) (Fig. 2).
Para los casos PCE-4 y PCE-5 la actividad de URO III-S reveló que la capacidad de la enzima para formar el isómero III estaba disminuida entre un 25-44% (Fig. 3).
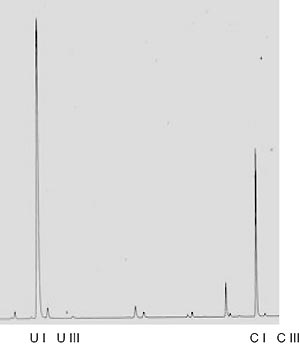
Figura 1. Patrón de excreción de porfirinas urinarias de la paciente PCE-3.
Tiempos de retención: URO-I: 5,9; URO-III: 6,3; COPRO-I: 15,3; COPRO-III: 15,9.
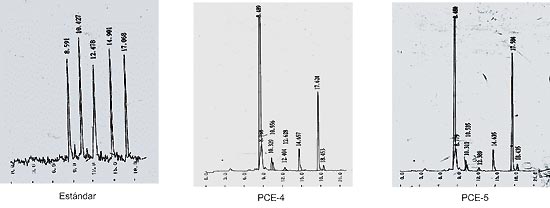
Figura 2: Porfirinas urinarias por HPLC.
Tiempos de retención: URO-I: 8,5; URO-III: 8,8; COPRO-I: 17,6; COPRO-III: 18,4.
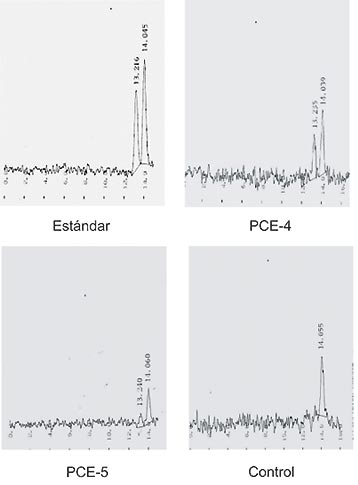
Figura 3. Formación de porfirinas por la URO III-S.
Tiempo de retención: URO I: 13,2´URO III: 14,0.
En la tabla se indican los porcentajes formados por la enzima en ambos pacientes.
Tabla IV. Evolución del paciente PCE-5.
DISCUSIÓN Y CONCLUSIONES
Las porfirias son enfermedades metabólicas de baja incidencia, 0,5 a 10 por 100.000 habitantes en diferentes poblaciones (5). Dentro de ellas la PCE, porfiria autosómica recesiva, es una de las menos frecuentes, habiéndose descripto alrededor de 200 casos a nivel mundial. Del análisis de estos casos surge que existe una relación genotipo-fenotipo que da lugar a presentaciones clínicas altamente variables que pueden ir desde casos muy severos, transfusión-dependiente, a casos de manifestación tardía sólo con leves lesiones cutáneas (22)(33).
La marcada deficiencia enzimática en esta porfiria a nivel de la URO III-S trae como consecuencia la producción aumentada y acumulación en glóbulos rojos de elevadas cantidades de porfirinas de la serie I que causan hemólisis y así las porfirinas circulantes son las responsables, por su alta foto-reactividad, de las graves lesiones cutáneas que presentan algunos de estos pacientes, pudiendo llegar hasta la mutilación principalmente en manos, dedos y cara. Además, las porfirinas se acumulan en huesos y dientes produciendo la típica eritrodoncia generalmente observada en estos pacientes.
Desde el punto de vista bioquímico, una de las características de esta porfiria es la excreción masiva de porfirinas por orina pudiendo encontrarse valores que superan en dos órdenes de magnitud el valor normal, principalmente intermediarios de la serie I y con predominio de la porfirina más hidrosoluble, la uroporfirina.
En este trabajo se describen 5 casos de PCE en este país, 4 en niños y uno de manifestación tardía que representan aproximadamente un 2% de los casos reportados en la literatura mundial
Todos los pacientes presentaron contenidos elevados de porfirinas en orina, sangre y materia fecal. El índice de porfirinas en plasma, que es un indicador rápido y simple para la identificación de las diferentes porfirias, se encontró también muy elevado en todos los casos, típico de una porfiria cutánea, que de acuerdo al máximo de emisión (619 nm) podría corresponder a una PCT o una PCE. El patrón de porfirinas urinarias confirmó el diagnóstico presuntivo de PCE o permitió, en el caso adulto, realizar el diagnóstico diferencial de porfiria.
Los casos descriptos incluyen diferentes grados de manifestación clínica. El más severo corresponde al PCE-3 que desde su nacimiento presentó hemorragias masivas, siendo así una paciente transfusión-dependiente que falleció a la edad temprana de 22 meses.
Los hermanos salteños (PCE-1 y PCE-2) representarían cuadros no tan severos pero con manifestaciones cutáneas serias y eritrodoncia, pese a lo cual actualmente se encuentran vivos y con buen estado de salud.
El PCE-4 constituye, dentro de las de manifestación temprana, una forma leve, ya que sus lesiones cutáneas no fueron importantes y no presentaba eritrodoncia.
En el año 2004 Ged y col. (33) describieron un caso similar. Una niña, perteneciente a una familia palestina con 4, de 13 hermanos, afectados y que pese a presentar una profunda deficiencia enzimática por ser una portadora homocigota de una mutación severa, sólo mostraba una porfirinuria moderada y sin sintomatología cutánea sugiriendo la presencia de un factor protector asociado a un posible gen modificador relacionado o no al camino biosintético del hemo (33).
El cuadro clínico y bioquímico del paciente PCE-4 comenzó a empeorar alrededor de los 5 años de edad y se decidió someterlo a un transplante de médula ósea. Según los estudios bioquímicos se produjo una disminución significativa de los valores de porfirinas en las diferentes muestras biológicas paralelamente a una mejoría clínica. Sin embargo, en el segundo control realizado, a un año del primero, se evidenció un incremento en los parámetros bioquímicos, principalmente en la excreción de porfirinas urinarias y el IPP, sin alteración de su mejoría clínica. Estas observaciones no coinciden con lo descripto por otros autores para transplantes realizados en niños, tanto de donantes compatibles relacionados como no relacionados, en los cuales se observó que los valores de porfirinas urinarias se mantuvieron constantes o en descenso entre 1 y 3 años post transplante (22)(34).
El último es un caso de manifestación tardía de los cuales sólo hay 13 descriptos mundialmente. Es por ello que inicialmente se sospechó que se trataba de una PCT, diagnóstico que se descartó al analizarse el patrón de excreción de porfirinas urinarias. El estudio se completó con la determinación de la actividad de la enzima deficiente en la PCT, la URO-D, cuyo valor de 16,84 U/mL GR resultó normal (VN: 16,35+3,56 U/mL GR). Es de hacer notar que, a pesar del elevado valor de porfirinas presentes en plasma y excretadas por orina, el paciente nunca tuvo manifestaciones cutáneas severas ni otras complicaciones. La explicación a este hecho queda aún por dilucidar.
La determinación de la actividad de la enzima deficiente en la PCE, URO III-S, en los glóbulos rojos de los pacientes PCE-4 y PCE-5, indicó que se encontraba disminuida en tan sólo un 25-44%, valor elevado para lo esperado para una enfermedad recesiva pero que estaría de acuerdo con la presentación clínica de la porfiria en estos pacientes.
Finalmente, es de destacar la importancia de un diagnóstico diferencial precoz de esta terrible enfermedad y de ser posible su genotipificación con el objetivo de, o bien aplicar el tratamiento y cuidados adecuados en los pacientes levemente afectados y evitar el agravamiento de sus lesiones, o en aquellos severamente afectados, decidir lo más tempranamente posible la posibilidad de un transplante de médula ósea que constituye hoy en día la única alternativa eficaz para brindarles una mejor calidad de vida.
Agradecimientos
El trabajo se llevó a cabo con subsidios del CONICET (2283/89) UBA (UBACYT X 195, X304) y la Agencia de Promoción Científica (PICT 059042). Se agradece al Dr. H. Muramatsu y a la Sra. V Castillo por la atención de los pacientes
Correspondencia
PROF. DR. A. M. DEL C. BATLLE
Viamonte 1881, 10 "A"
1056 CIUDAD AUTÓNOMA DE BUENOS AIRES, Argentina
Tel: (5411) 4812-3357, Fax: (5411) 4811-7447
E-mail: batlle@mail.retina.ar
Referencias bibliográficas
1. Batlle AM del C. Porfirias Humanas. Signos y Tratamientos. En: Actualizaciones Médico Bioquímicas. Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. La Plata: Federación Bioquímica de la Provincia de Buenos Aires; 1997. p. 37-82.
2. Romeo G, Levin EY. Uroporphyrin III cosynthase in human congenital erythropoietic porphyria. Proc Nat Acad Sci USA 1969; 63: 856-63.
3. Kappas A, Sassa S, Galbraith RA, Nordmann Y. The Porphyrias. In: Scriver CS, Beaudet al, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Diseases.7th ed. York: Mc Graw-Hill; 1995. p. 2.103.
4. Desnick RJ, Glass IA, Xu W, Solís C, Astrin KH. Molecular genetics of congenital erythropoietic porphyria. Semin Liver Dis 1998; 18: 77-84.
5. Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis. X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet al, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Diseases, 8th ed. New York: McGraw-Hill; 2001; p. 2.961-3.062.
6. Burton G, Fagerness PE, Hosozawa S, Jordan PM, Scott AI. 13C-NMR evidence for a new intermediate, preuroporphyrinogen, in the enzymic transformation of porphobilinogen into uroporphyrinogens I and III. J Chem Soc Chem Comunn 1979; 5: 202-4.
7. Jordan PM, Burton G, Nordlov H, Schneider MM, Pryde L, Scott AI. Preuroporphyrinogen: a substrate for uroporphyrinogen III cosyntethase. J Chem Soc Chem Comunn 1979; 5: 204-5.
8. Battersby AR, Fookes CJR, Marchand GWJ, McDonald E. Biosynthesis of the pigments of life: formation of the macrocycle. Nature 1980; 285: 17-21.
9. Bickers DR, MAP: The Porphyrias. In: Fitzpatrick TB, Eisen AZ, Wolf K, Freedberg IM, Austen KF (eds): Dermatology in General Medicine. New York: Mc Graw-Hill;1987. p. 1.679.
10. Astrin KH, Warner CA, Yoo HW, Goodfellow PJ, Tsai SF, Desnick RJ. Regional assignment of the human uroporphyrinogen III synthase (UROS) gene to chromosome 10q25.2-q26.1. Hum Genet 1991; 87: 18-22.
11. Xu W, Astrin HK, Desnick RJ. Molecular basis of congenital erythropoietic porphyria mutations in the human uroporphyrinogen III synthase gene. Hum Mutat 1996; 7: 187-92.
12. Desnick RJ, Astrin KH. Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol 2002; 117: 779-95.
13. Mc Goven MM, Anderson KE, Astrin KH, Desnick RJ. Inherited porphyries. In: Rimoin DL; Connor JM, Pyeritz RE (eds). Emery and Rimmoin´s. Principles and Practice of medical genetics, vol 2 (3rd ed.). New York: Churchill-Livingstone; 1996. p. 2.009.
14. Seip M, Thune PO, Eriksen L. Treatment of photosensitivity in congenital erythropoietic porphyria with b-carotene. Acta Derm Veneorol (Stockholm) 1974; 54: 239-40.
15. Pimstone NR, Gandhi SN, Mujerki SK. Therapeutic efficacy of oral charcoal in congenital erythropoietic porphyria. N Engl J Med 1987; 316: 390-3.
16. Piomelli S, Poh-Fitzpatrick MB, Seaman C, Skolnick LM, Berdon WE. Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with high-level transfusions. N Engl J Med 1986; 314: 1.029-31.
17. Guarini L, Piomelli S, Poh-Fitzpatrick MB. Hydroxyurea in congenital erythropoietic porphyria. N Engl J Med 1994; 330: 1.091-2.
18. Kauffman L, Evans DI, Stevens RF, Weinkove C. Bone marrow transplantation for congenital erythropoietic porphyria. Lancet 1991; 337: 1.510-1.
19. Zix-Kieffer I, Langer B, Eyer D, Acar G, Racadot E, Schlaeder G, et al. Successful cord blood stem cell transplantation for congenital erythropoietic porphyria (Günther disease). Bone Marrow Transplant 1996; 18: 217-20.
20. Thomas C, Ged C, Nordmann Y, de Verneuil H, Pellier I, Fischer A, et al. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr 129: 453-6.
21. Lagarde C, Hannel-Tellac D, De Prost Y, Blanche S, Thomas C, Fischer A, et al. Allogenic bone marrow transplantation in congenital erythropoietic porphyria. Günther disease. An Dermatol Venereol 1998; 125: 114-7.
22. Tezcan I, Xu W, Gurgey A, Tuncer M, Cetin M, Oner C, et al. Congenital Erythropoietic Porphyria successfully treated by allogenic bone marrow transplantation. Blood 1998; 92: 4.053-8.
23. Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol 2001; 45: 279-82.
24. Shaw PH, Mancini AJ, Mc Connell JP, Brown D, Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogenic stem cell: a case report and a review of the literature. Bone Marrow Transplant 2001; 27: 101-15.
25. Kontos AP, Ozoq D, Bichakjian C, Lim HW. Congenital erythropoietic porphyria associated with myelodysplasia presenting in a 72-year-old man: report of a case and review of the literature. Br J Dermatol 2003; 148: 160-4.
26. Magnin PH, Batlle AM del C, Lenczner JM, Parera VE, Stella AM, Godoy O. Porfiria congénita eritropoyética en dos niños. Estudio clínico y bioquímico. Rev Argent Dermatol 1986; 67: 163-70.
27. Melito VA, Rossetti MV, Parera VE, Batlle A. Non frequent porphyrias in the Argentinean population. Rev Argent Dermatol 2007 (en prensa).
28. Batlle AM del C. El Laboratorio de las Porfirias. En: Actualizaciones Médico Bioquímicas. Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. La Plata: Federación Bioquímica de la Provincia de Buenos Aires; 1997. p.145 -71.
29. Tsai SF, Bishop DF, Desnick RJ. Purification and properties of uroporphyrinogen III synthase from human erythrocytes. J Biol Chem 1987; 262: 1.268-73.
30. Lim CK, Rideout JM, Wright DJ. High-performance liquid chromatography of naturally occurring 8-, 7-, 6-, 5- and 4-carboxylic porphyrin isomers. J Chromatogr 1983; 282: 629-41.
31. Falk JE, Benson A. Separation of uroporphyrin esters I and III by paper chromatography. Biochem J 1953; 55: 101-4.
32. Afonso SG, Chinarro S, Stella AM, Lenczner JM, Batlle AM del C, Magnin PH. Uroporfirinógeno decarboxilasa eritrocitaria y hepática en porfiria cutánea tardía. Rev Argent Dermatol 1985; 66: 12-23.
33. Ged C, Megarbane H, Chouery E, Lalanne M, Megabarne A, de Verneuil H. Congenital erythropoietic porphyria: report of a novel mutation with absence of clinical manifestations in a homozygous mutant sibling. J Invest Dermatol 2004; 125: 589-91.
34. Dupuis-Girod S, Akkari V, Ged C, Galambrun C, Kebaili K, Deybach JC, et al. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Günther disease). Eur J Pediatr 2005; 164: 104-7. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Aceptado para su publicación el 8 de junio de 2007

