










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. v.41 n.4 La Plata oct./dic. 2007
BIOLOGÍA MOLECULAR
Marcadores genéticos pronósticos en la infección por el hiv-1*
Genetic factors in hiv-1 infection
Andrea María Mercedes Mangano1
1. Investigadora Adjunta de la Carrera del Investigador Científico del Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). Laboratorio de Biología Celular y Retrovirus. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan". Dra. de la Universidad de Buenos Aires, Área de Microbiología. Profesora Adjunta, Cátedra de Microbiología, Departamento de Microbiología, Inmunología y Biotecnología de la Facultad de Farmacia y Bioquímica de la Universidad de Buenos Aires.
* Laboratorio de Biología Celular y Retrovirus. Hospital de Pediatría "Prof. Dr. Juan P. Garrahan". Buenos Aires, Argentina.
Premio Bienal FABA 2006
Resumen
En la última década una serie de descubrimientos ha permitido caracterizar las complejas interacciones virus-hospedador que determinan la patogenia de la infección por el HIV-1. El ingreso del HIV a la célula es el primer paso en el establecimiento de la infección viral. En esta etapa, el virus debe interaccionar con dos tipos de receptores en la superficie celular, el CD4 y un receptor de quimioquina (principalmente CCR5 o CXCR4). La utilización de los co-receptores celulares estará determinada por las características de la envoltura viral, por ello los virus que utilizan el CCR5 o el CXCR4 son denominados R5 y X4 respectivamente. Las interacciones virus - co-receptor como ser R5/CCR5 pueden ser bloqueadas, al menos parcialmente, por las quimioquinas, que son ligandos fisiológicos de los receptores como CCL3, CCL4 y CCL5. Varios estudios han demostrado que variaciones genéticas tanto en los co-receptores como en las quimioquinas pueden condicionar la transmisión del HIV-1 y la evolución de la enfermedad. En este artículo se realiza una revisión de la participación de factores virales y celulares en la inmunopatogenia de la infección por el HIV-1 y de la experiencia de la autora en Argentina.
Palabras clave: Virus de la inmunodeficiencia humana; Co-receptor; Quimioquinas; Factores genéticos; Patogenia; Síndrome de inmunodeficiencia infantil
Summary
In the last decade numerous findings have helped to unveil the complex viral-host interactions determining HIV-1 pathogenesis. Viral entry is the first step in the establishment of HIV infection. In this phase, the virus needs to interact with two cellular receptors, the CD4 molecule and a chemokine receptor (mainly CCR5 or CXCR4). Viral envelope features determine the usage of cellular coreceptors thus, virus that use CCR5 or CXCR4 are named R5 and X4, respectively. Viral-coreceptor interactions could be blocked, at least partially, by the CCL3, CCL4 and CCL5 chemokines. Several studies have shown that genetic variations in the chemokines and their receptors may affect HIV-1 transmission and disease progression. In this article the influence of host genetic and viral factors is reviewed as well as our experience in Argentina.
Key words: Human immunodeficiency virus; Coreceptor; Chemokine; Host genetic factors; Pathogenesis; Pediatric immunodeficiency syndrome
Historia y origen de la infección
A principio de la década del 80 se describió una nueva entidad clínica que se conoció como el síndrome de inmunodeficiencia adquirida (SIDA) (1). Los primeros casos se registraron en las ciudades de San Francisco y Nueva York (EE.UU.), y se caracterizaban por presentar infecciones oportunistas, principalmente neumonía por Pneumocystis carinii o tumores sólidos como el sarcoma de Kaposi, que en forma completamente inusual se presentaban en dos grupos distintos: varones homosexuales y jóvenes adictos a drogas por vía endovenosa (2-5). Todos los individuos afectados presentaban una disfunción progresiva e irreversible de la inmunidad celular. Más tarde se sumaron casos de SIDA en hemofílicos, hemotransfundidos, luego mujeres que eran parejas de hombres con SIDA y finalmente en niños (1)(6-8). Los datos epidemiológicos acumulados sugerían que un agente infeccioso estaba involucrado como causante de la inmunodeficiencia y que la transmisión se producía fundamentalmente por vía sexual, parenteral y vertical.
Durante 1983, tres virus fueron propuestos como agentes etiológicos del SIDA: el LAV (lymphoadenopathy associated virus) (9), el HTLV III (human T lymphotropic virus type III) (10)(11) y el ARV (AIDS related virus) (5). La comparación de las secuencias nucleotídicas de los distintos aislamientos virales demostró que pertenecían a un único virus (12) que el Comité Internacional de Taxonomía de Virus denominó Virus de la Inmunodeficiencia Humana Adquirida "HIV" (Familia Retroviridae, subfamilia Lentiviridae). En 1985 en África occidental se aisló un retrovirus similar al inicialmente descripto aunque su secuencia genética difería en aproximadamente un 30% del HIV. Se lo consideró un tipo diferente y se lo denominó HIV tipo 2 (HIV-2) (13) para diferenciarlo del primero, que fue designado HIV tipo 1 (HIV-1).
Los dos tipos de HIV (HIV-1 y HIV-2) que causan SIDA en humanos representan infecciones zoonóticas transmitidas por lentivirus de primates (14)(15). El primer reservorio identificado como hospedador del HIV-2 fue el primate sooty mangabey (Cercocebus atys) (16). Posteriormente, se demostró que el HIV-1 provendría del virus de la inmunodeficiencia simiana que alberga el chimpancé Pan troglodytes (SIV cpz) (14). Por análisis genéticos del ADN mitocondrial se demostró que las tres cepas de SIV cpz más relacionadas con el HIV-1 provendrían de una única subespecie, la Pan troglodytes troglodytes, que habita en la región central de África donde se acepta que surgió el SIDA (14).
Actualmente se reconocen tres grupos genotípicamente diferentes del HIV-1: el grupo M (main), el grupo O (outlier) y el grupo N (new). El grupo M (17) se ha diseminado a nivel mundial y se han descripto 9 subtipos (A-D, F-H, J y K) y diversas formas recombinantes. Analizando la secuencia del HIV en una muestra extraída de un congolés en 1959 se pudo demostrar que la diversificación de todos los subtipos del grupo M ocurrió muy recientemente, en los últimos 50 años (18) y se estima su origen alrededor de 1930, pudiendo extenderse de 1910-1950. Los análisis de evolución de los tres grupos del HIV-1 (M, N y O) al ser comparados con los aislados de SIV cpz, avalan que estos tres grupos representan tres transferencias independientes del SIV cpz a la población humana.
Desde la aparición del SIDA, en menos de 10 años, la infección por el HIV se transformó en una pandemia. El HIV-1 es el responsable de la diseminación mundial y en la actualidad constituye uno de los problemas de salud más serios y preocupantes. El HIV-2 no se ha diseminado con la velocidad del HIV-1, es endémico de África central y es el agente causal de una forma de SIDA más leve que la causada por el HIV-1, sugiriendo una menor transmisibilidad y patogenicidad. En la mayoría de las regiones del mundo se han diagnosticado casos de SIDA y la diseminación del HIV-1 es mayor en las áreas con una gran densidad poblacional. Según las últimas estimaciones del Programa Conjunto de las Naciones Unidas sobre HIV/SIDA (ONUSIDA) y de la Organización Mundial de la Salud (OMS), se estima que el número de personas infectadas con el HIV-1 es de 39,6 millones (33,4 a 46 millones) y ya han fallecido más de 20 millones (19).
Curso de la infección viral
El curso de la infección por el HIV-1 se puede dividir en tres fases: 1) infección aguda primaria; 2) período de latencia clínica o fase crónica de la infección; y 3) fase sintomática del SIDA.
El período de infección primaria o de primoinfección se caracteriza por una activa replicación viral, como también por el surgimiento de una respuesta inmunológica específica contra el HIV-1 y el control de la viremia. Esta infección aguda puede extenderse de cuatro a ocho semanas abarcando dos etapas. Una primera etapa de diseminación viral que ocurre durante las tres o cuatro primeras semanas posteriores a la infección y culmina con el pico de viremia. En este período no se detectan anticuerpos específicos contra el HIV-1 y por eso se denomina período ventana. Durante las primeras semanas se observa un descenso transitorio en el número absoluto de linfocitos T CD4+ en sangre periférica y altos niveles de carga viral plasmática (20). La segunda etapa se caracteriza por el descenso brusco de la viremia que se correspondería con la eliminación parcial del virus en sangre (21) concomitantemente con la detección de anticuerpos específicos contra el virus, o sea con el período de seroconversión, que acompaña un ascenso de los linfocitos T CD4+. En la Figura 1 se ilustra el curso típico de la infección por HIV-1. Durante la infección primaria, el 50% de los sujetos infectados pueden presentar síntomas clínicos que semejan una mononucleosis infecciosa que, cuando se prolonga, se asocia con un pronóstico desfavorable de la enfermedad. La respuesta humoral específica aparece entre las 2 semanas y 3 meses posteriores a la infección.
Después de la seroconversión, sobreviene el período de latencia clínica o fase crónica, que antecede al inicio de la enfermedad y puede variar entre 1 a 15 años o más. Durante todo este período, el sujeto infectado permanece libre de síntomas y signos marcadores del SIDA. Sin embargo, el curso de la infección continúa de manera inadvertida y se suceden una serie de fenómenos virológicos e inmunológicos que se acompañan de deterioro gradual e inexorable del sistema inmunitario. Comienza una reducción lenta y progresiva del número absoluto de linfocitos T CD4+ que, en general, persiste durante años. En algunos casos, la reducción del número de estas células ocurre en forma acelerada y conduce a la aparición de SIDA temprano.
Las etapas finales de la enfermedad se caracterizan por presentar una inmunosupresión severa con recuentos de linfocitos T CD4+ muy bajos y altos niveles de viremia plasmática los cuales aumentan con las infecciones oportunistas concomitantes que conducen finalmente a la muerte.
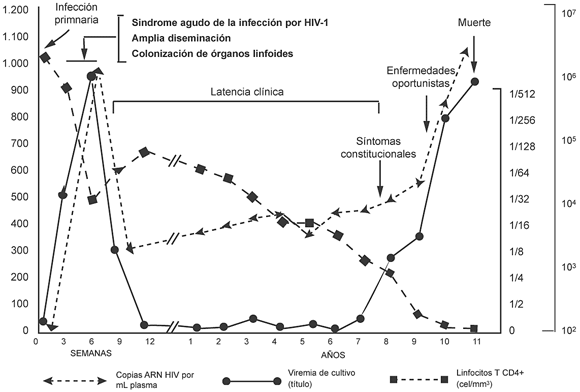
Figura 1. Curso de la infección por HIV-1. Se muestra el perfil inmunológico,
y virológico según la fase clínica de la infección. Adaptado de Pantaleo et al (20).
Patogenia del HIV-1
El HIV-1 ataca directamente al sistema de defensas del hombre infectando a las células del sistema inmune que incluye linfocitos T, macrófagos, y células dendríticas, con una replicación más eficiente cuando las células están activadas. Por ello, los eventos que se producen para generar una respuesta específica contra el HIV-1 irónicamente exponen al sistema inmune a ciclos repetidos de activación, favoreciendo por lo tanto la infección y destrucción de sus propias células (20).
Luego de la infección primaria, la reducción en la viremia plasmática y la aparición de anticuerpos específicos contra el HIV-1 van acompañadas por un ascenso de linfocitos T CD4+. Esto demuestra que frente a la infección viral, la respuesta inmune del hospedador es importante y eficiente, aunque no suficiente para la erradicación viral.
En el organismo infectado la protección contra la infección estaría mediada por los anticuerpos específicos contra el HIV-1 (22), que eliminarían los viriones de circulación, los linfocitos T citotóxicos específicos responsables de la eliminación de las células infectadas, y las células asesinas naturales o "natural killers" que son capaces de reconocer células infectadas (23).
Durante la evolución de la enfermedad se producen alteraciones en los distintos tipos celulares que participan en la respuesta inmune y principalmente en la subpoblación de linfocitos T CD4+, cuya depleción es la principal responsable de la profunda inmunosupresión que caracteriza al síndrome. La destrucción de células T CD4+ puede ocurrir por acción directa del virus y en forma indirecta a través de mecanismos inmunológicos. Aún hoy está en discusión cuál de los dos mecanismos es el preponderante, aunque seguramente ambos mecanismos contribuyen en la patogenésis del HIV-1 y tal vez el predominio de uno sobre el otro varíe de acuerdo al período de la infección viral (25).
El efecto citopatógeno del HIV puede producirse por destrucción de la célula infectada por dos mecanismos distintos: promoviendo la fusión entre células infectadas y no infectadas formando sincicios celulares, y provocando la muerte celular individual por mecanismos hasta el presente poco conocidos. Se ha sugerido que la destrucción de la célula infectada puede ocurrir directamente por activación de la apoptosis o indirectamente a través de un mecanismo parácrino mediante la liberación de factores virales que actuarían sobre las células circundantes a la infección viral (25)(26).
Por otro lado, la alteración en la producción de citoquinas por parte de los linfocitos T CD4+ contribuiría a la disfunción de otras células, principalmente las efectoras, como los linfocitos T CD8+ pero también en células NK, linfocitos B, y algunas funciones de macrófagos (27)(28).
Durante la infección también se observa una activación de los linfocitos T y B (20). Durante la fase asintomática, los linfocitos del tejido linfoide son los protagonistas de la enfermedad. En los ganglios linfáticos es donde tiene lugar el secuestro vírico, la activación celular y la depleción de los linfocitos T CD4+. Las células dendríticas, a través de sus receptores Fc, captan células infectadas y viriones ligados a inmunoglobulinas específicas y los procesan para más tarde presentarlos de nuevo a células B memoria, manteniendo la respuesta de anticuerpos. En los ganglios linfáticos se observa una hiperplasia folicular y expansión de la red que forman las células dendríticas foliculares, indicativo de activación del tejido linfoide. Paradójicamente, la activación crónica de los linfocitos T CD4+ en los ganglios linfáticos favorece la infección y diseminación viral. Conforme avanza la enfermedad, hay un progresivo agotamiento del mecanismo de secuestro de los linfocitos T CD4+ por parte de las células dendríticas que se asocia con una mayor replicación viral.
En conclusión, el SIDA involucra un proceso de inmunoactivación que siendo beneficioso al principio, tiene efecto contraproducente cuando se hace crónico. Esta hiperactivación, potenciaría la replicación viral y activaría los mecanismos de muerte celular programada, pero también provocaría una desregulación con respuestas inmunes aberrantes (20). Todo esto conduciría a un agotamiento del sistema inmune provocando una fuerte inmunosupresión característica de los estadios tardíos, con destrucción del tejido linfoide que favorecería la diseminación del HIV-1 y, finalmente, con el colapso de la respuesta inmune disminuyen todas las subpoblaciones linfocitarias aumentando la susceptibilidad a adquirir infecciones oportunistas y/o tumores además de una fuerte caída de los anticuerpos específicos contra el virus (29)(30).
Marcadores bioquímicos pronósticos de la infección
El recuento de linfocitos T CD4+ es uno de los marcadores de laboratorio más empleados para el seguimiento de la evolución de la infección ya que su reducción refleja el grado de inmunodeficiencia del paciente infectado.
Los valores de linfocitos T CD4+ se utilizan para definir el estadio inmunológico y para recomendar el momento preciso para el inicio y monitoreo de la terapia antirretroviral específica. Sin embargo, su valor predictivo es relativo ya que el descenso brusco de linfocitos T CD4+ generalmente precede en poco tiempo a la aparición de síntomas marcadores de la inmunodeficiencia severa.
Desde que se reconoció que la replicación del HIV-1 ocurre en todos los estadios de la infección como un proceso continuo y dinámico, con una producción diaria de más de 1.000 millones de viriones, se ha tratado de cuantificar la viremia plasmática. A partir de 1995, la medición de la carga viral plasmática medida como copias de ARN viral/mL de plasma se considera el marcador más confiable como predictor de la evolución de la infección y como un indicador para el inicio y el control terapéutico específico (31). Sin embargo, el recuento de linfocitos T CD4+, como indicador del estado de inmunodeficiencia existente, es un marcador útil y complementario a la carga viral plasmática.
Factores virales y celulares en la inmunopatogenia de la infección por el HIV-1
Desde el inicio de la epidemia causada por HIV se ha observado que frente a la exposición viral reiterada hay individuos que no se infectan, indicando una evidente protección natural. Por ejemplo, niños no infectados nacidos de madres HIV-seropositivas o individuos con exposiciones reiteradas al HIV por tener parejas HIV-seropositivas. Además, en los sujetos HIV-infectados existe una gran variabilidad individual en cuanto a la morbi-mortalidad de la infección. Algunos pacientes infectados desarrollan SIDA en forma muy rápida, llamados progresores rápidos; por el contrario, algunos pacientes HIV-1 seropositivos permanecen asintomáticos por más de una década sin tratamiento antirretroviral, denominados progresores a largo plazo -LTNP (long term non progressors)-. Por ello, desde los inicios de la epidemia se trata de dilucidar por qué algunos individuos presentan baja susceptibilidad a la infección por el HIV-1. Entre los factores que pueden contribuir a la "resistencia" a la infección por el HIV se encuentran los factores inherentes al virus, como ser exposición a una cepa menos patogénica, y factores del hospedador, por ejemplo debido a una predisposición genética contra la infección o la presencia de algún factor celular protector.
Desde el punto de vista fenotípico, una de las características del HIV-1 es su gran variabilidad respecto de las propiedades biológicas, como la capacidad de formar sincicios celulares, el índice de replicación, y el tropismo celular, que podrían contribuir a las variaciones observadas en el curso clínico de la infección.
De acuerdo con el tropismo celular se divide a las cepas del HIV-1 en dos categorías: macrófago-trópicas (M-trópicas) y T-trópicas (32-34). Las cepas M-trópicas son las que infectan macrófagos y linfocitos T primarios pero no son capaces de inducir la formación de sincicios in vitro, o sea son no inductoras de sincicios celulares (NSI). En general, las cepas NSI se pueden aislar durante todo el curso de la infección (35), en general son de replicación lenta (36), y son siempre las predominantes en la infección primaria, por ello se considera que tendrían un mayor poder de transmisión independientemente de la vía de contagio (37). Las cepas T-trópicas infectan linfocitos T primarios y de líneas celulares T establecidas y son las que inducen la formación de sincicios celulares (SI) sobre las células T CD4+ de la línea celular MT-2 (Fig. 2). Las cepas Si se aíslan predominantemente en pacientes con una rápida progresión clínica y en estadios tardíos de la infección viral (33). Las variantes T-trópicas emergerían con el avance de la enfermedad acelerando la inmunosupresión en el 50% de los adultos infectados (37)(38). En algunos casos el cambio fenotípico NSI-SI coincide con un rápido aumento en la carga viral plasmática y disminución en el recuento de linfocitos T CD4+. También se han identificado algunas cepas que pueden infectar macrófagos, linfocitos y líneas celulares T establecidas, son las consideradas dual-trópicas y son SI.
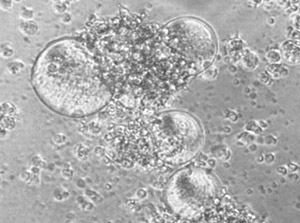
Figura 2. Formación de sincicios celulares producido por el HIV-1 sobre la línea celular MT-2. Observación al microscopio óptico invertido con un aumento de 16X.
Desde el punto de vista genotípico, el virus se encuentra en el huésped como una mezcla de variantes genéticas estructuralmente relacionadas, que se denominan cuasiespecies. Los retrovirus al replicarse y debido a la gran cantidad de errores cometidos por la transcriptasa inversa (aproximadamente un error por ciclo), generan una serie de variantes heterogéneas. Las más homogéneas y frecuentes constituyen lo que se llama cepa "master", que va a caracterizar a la cuasiespecie. Además, la recombinación entre genomas virales similares pero diferentes, que pueden co-existir en células infectadas, aumenta la diversidad genética. En América, las recombinantes más frecuentemente hallados son las B/F (39)(40). La gran variabilidad y su continua evolución son responsables de la rápida aparición de variantes resistentes a la neutralización por anticuerpos, a los linfocitos T citotóxicos y a las drogas usadas en la terapia antiviral.
Todo esto hace que del fenómeno de la variabilidad deriven características epidemiológicas de gran importancia, tales como la transmisión y circulación viral, progresión de la enfermedad, especificidad celular, resistencia a los antirretrovirales y dificultades para el desarrollo de vacunas.
Además de la heterogeneidad viral, las características propias del hospedador también condicionan la susceptibilidad al HIV-1. Poco después del aislamiento del HIV se identificó a la molécula de CD4 como su receptor primario. Estudios in vitro demostraron que en células de ratón transfectadas con CD4 ocurría adhesión viral pero no ingreso del virus a la célula sugiriendo la necesidad de un cofactor/es celulares para que ocurra la infección viral. Esto, sumado a la existencia de cepas del HIV-1 con distinto tropismo celular, M-trópicas y T-trópicas, sugería la posibilidad de que estas cepas utilizaran complementariamente co-receptores distintos (41).
Luego de una década de intensa búsqueda, en 1996 se descubrió que las moléculas en cuestión eran receptores específicos de quimioquinas que actúan como co-receptores del HIV-1 (42)(43). Uno de los estudios iniciales fue el trabajo de Paxton, et al. (44) que demostró que las células de individuos expuestos al HIV no infectados presentaban cierta resistencia a la replicación viral y, que en especial los linfocitos T CD4+ de dos de estos individuos eran totalmente refractarios a la replicación de cepas HIV-1 M-trópicas pero no de virus T-trópicos. Estas observaciones fueron seguidas por dos estudios que condujeron a la caracterización de los co-receptores del HIV-1. Uno de ellos fue la identificación de las b-quimioquinas: MIP-1a (proteína inflamatoria de macrófagos 1a), MIP-1b (proteína inflamatoria de macrófagos 1b) y RANTES (regulation on activation, normal T cell expressed and secreted), como los factores solubles secretados por las células T CD8+ capaces de inhibir la replicación in vitro del HIV-1 (45). En personas expuestas no infectadas se detectaron altos niveles de estas quimioquinas asociándose con una protección "natural" a la infección (44)(47). Estudios posteriores mostraron que el efecto antiviral de estas quimioquinas era a nivel de la entrada del virus y que la inhibición de la infección estaba restringida sólo a las cepas M-trópicas (41)(47)(48).
El otro estudio fue el realizado por Feng, et al. (43) que condujo a la identificación del primer co-receptor del HIV-1. Esta molécula inicialmente denominada fusina, en presencia del CD4, permite la fusión de membranas mediada por virus T-trópicos y M-trópicos. Su ligando fisiológico fue rápidamente descripto como el factor derivado del estroma tipo 1 (SDF-1) (49)(50).
El SDF-1 es una quimioquina de tipo CXC o a cuya acción es modular la función de los linfocitos B y de las células progenitoras hematopoyéticas. Además, tiene la capacidad de inhibir específicamente la replicación de los aislados T-trópicos del HIV-1, probablemente por bloqueo o regulación en menos (downregulation) del receptor. Por las características de su ligando natural, la fusina fue redesignada como el receptor CXCR4.
Estos hallazgos condujeron a que, poco después y en forma independiente, 5 grupos demostraran que el receptor celular de quimioquinas CCR5 es el principal co-receptor de los virus M-trópicos, y que las b-quimioquinas RANTES, MIP-1a y MIP-1b son sus ligandos naturales (41)(47)(48)(51)(52).
Las quimioquinas son un grupo de citoquinas de bajo peso molecular (8-10 kDa), capaces de atraer leucocitos hacia los sitios de inflamación. Ya se han descripto más de 40 quimioquinas que de acuerdo con el número y distancia de las cisteínas conservadas en los extremos aminoproximales se clasifican en 4 clases: CXC (a), CC (b), C (g) y CX3C (d). La mayoría de las quimioquinas identificadas pertenecen a los grupos de tipo CC o CXC. Actualmente las quimioquinas se designan como CCL: RANTES es denominada CCL5, MIP-1a como CCL3 y MIP-1b como CCL4.
Las quimioquinas producen sus efectos a través de la interacción con los receptores celulares específicos. Estos receptores pertenecen a la superfamilia de receptores con 7 dominios transmembrana que acoplan proteína G (GPCR) (53). La interacción de las quimioquinas con sus receptores es bastante promiscua. La mayoría de los receptores interactúan con múltiples ligandos y a su vez la mayoría de los ligandos actúa sobre más de un receptor aunque generalmente de una misma clase. Esta propiedad fue utilizada para clasificar a los receptores.
Además de los co-receptores principales del HIV-1, CCR5 y CXCR4, in vitro se han descripto otros receptores que pueden ser utilizados como co-receptores de entrada para el virus, aunque generalmente con menor eficiencia. Entre ellos se encuentran el CCR2, CCR3, CCR8, CCR9, CX3CR1, y varios receptores huérfanos como el STRL 33 ("Bonzo"), Gpr15 ("Bob"), Gpr1, APJ y Chem 223 (54)(55). Los receptores de quimioquinas son expresados por distintos tipos celulares incluyendo leucocitos, células endoteliales, neuronas, células epiteliales y células de la microglia del cerebro.
La relación entre el fenotipo viral y la utilización de co-receptores condujo a la creación de una nueva clasificación para las cepas de HIV-1. Las variantes M-trópicas consideradas transmisoras son denominadas R5, y las T-trópicas que son aisladas de pacientes con rápida progresión clínica y en estadios tardíos de la infección viral son denominadas X4 (Fig. 3) (56). La mayoría de las variantes que emplean el CXCR4 también pueden utilizar el CCR5 y son las denominadas dual trópicas o R5X4.
El descubrimiento de los co-receptores del HIV permitió profundizar los estudios a nivel del mecanismo de entrada del virus. La unión de la proteína de envoltura con el receptor celular CD4 induce cambios conformacionales en la subunidad gp120. La interacción del complejo gp120-CD4 con el co-receptor apropiado es la que permite los cambios conformacionales finales en la proteína de envoltura que provoca la exposición de la secuencia fusogénica gp41 que conduce a la fusión de membranas y la consecuente entrada viral (57).
El hecho que en individuos expuestos al HIV-1 no infectados se detectaron altos niveles de quimioquinas, orientó el estudio de las quimioquinas y sus receptores como mediadores en la patogénesis del HIV-1. Las quimioquinas pueden presentar un efecto paradójico. Por un lado, pueden bloquear la entrada del HIV induciendo la internalización del co-receptor en endosomas que previenen la formación del complejo gp120-CD4-co-receptor, y por lo tanto, la fusión de membranas y la infección viral (57). Por otra parte, inducen una respuesta inflamatoria atrayendo células blanco no infectadas al sitio de replicación viral activa, e inducen señalización intracelular que aumenta la replicación del HIV-1.
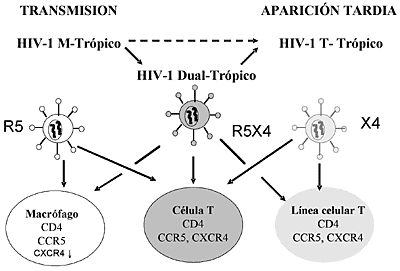
Figura 3. Tropismo celular del HIV-1. Las cepas de HIV-1 M-trópicas, R5 infectan macrófagos y constituyen las variantes transmisoras. A medida que avanza la infección, en la mitad de los pacientes infectados puede ocurrir un cambio de tropismo, con el surgimiento de cepas T-trópicas X4 que pueden infectar células y líneas celulares T. Las variantes dual-trópicas R5X4 son consideradas variantes de transición entre las R5 y las X4.
Variaciones genéticas en los co-receptores condicionan la susceptibilidad al HIV-1
Varias investigaciones se iniciaron para estudiar el rol de las quimioquinas y sus receptores en la biología y patogenia del HIV-1. Poco después del descubrimiento del CCR5 como el principal co-receptor del HIV-1, a mediados de 1996 tres grupos independientes, estudiando las células de individuos expuestos no infectados, identificaron una deleción de 32 pb en la región codificante del gen del CCR5 (CCR5-D32), que provoca la aparición de un codón de terminación prematuro, y genera una proteína truncada incapaz de ser expresada en la superficie celular (58-60). Los individuos que portan este defecto en forma homocigota (CCR5-D32/D32) serían "naturalmente resistentes" a la infección, ya que la ausencia del receptor CCR5 en la superficie celular impide la entrada de cepas R5, consideradas las variantes transmisoras. La deleción CCR5-D32 es casi exclusiva de poblaciones caucásicas con 1% de homocigotas, y 10-20% heterocigotas (61). Varios estudios epidemiológicos demostraron la ausencia del genotipo homocigota CCR5-D32/D32 en más de 5.000 individuos infectados. Sin embargo, el reporte de 8 casos de individuos con genotipo CCR5-D32/D32 que contrajeron la infección demostró que esta resistencia no es absoluta (62-67). En algunos casos se pudo comprobar que las cepas aisladas utilizaban exclusivamente el CXCR4 sugiriendo que los virus X4 podrían iniciar la infección, aunque no se pudo descartar que haya ocurrido un cambio en el fenotipo en las etapas iniciales de la infección. La baja frecuencia con que ocurren estos casos demuestra que el alelo CCR5-D32 confiere una clara ventaja protectora y que los virus que utilizan el CCR5 son los predominantemente involucrados en la transmisión del HIV-1. En individuos no infectados con alta exposición al HIV-1 se halló elevada la frecuencia de homocigoctas CCR5-D32/D32 (hasta 5%) apoyando el rol protector ejercido por este genotipo.
Los individuos adultos heterocigotas CCR5+/D32 se encuentran con más frecuencia en cohortes de pacientes LTNP y en individuos expuestos no infectados (58-60)(68). Independientemente de la vía de ingreso, la expresión heterocigota CCR5+/D32 no afecta la transmisión viral (59)(60)(69)(70-72). Sin embargo, en individuos adultos HIV-1 infectados se encontraron menores niveles de carga viral plasmática, menor tasa de disminución de linfocitos T CD4+ y períodos mayores libre de enfermedad con un retraso promedio de 2 a 4 años en el desarrollo de SIDA (68)(72).
La distribución poblacional del alelo CCR5-D32 es ampliamente variable de acuerdo con el origen étnico: es más frecuente en poblaciones caucásicas y está prácticamente ausente en poblaciones africanas, asiáticas y aborigen-americanas (73-75). En individuos caucásicos, la frecuencia alélica es aproximadamente del 8%, con 1% de homocigotas y entre 10% a 20% de heterocigotas. Entre las poblaciones europeas se registraron diferencias en la distribución geográfica de la mutación con un gradiente de norte a sur, desde los alrededores del mar Báltico (20%) hacia la costa mediterránea (5%). De acuerdo con estudios de evolución, se estima que la mutación CCR5-D32 probablemente apareció en un evento único y relativamente reciente en la población finno-húngara, con una edad de la mutación estimada en aproximadamente 1.400 años (375-3.675) (76)(77). Esto sugiere que la mutación apareció luego de la separación de las poblaciones europeas y asiáticas y por ello no se encuentra en poblaciones asiáticas o africanas.
Las variaciones genéticas en otros co-receptores como el co-receptor secundario CCR2 también pueden modificar la susceptibilidad a la infección por el HIV-1. En el gen del CCR2 se identificó un polimorfismo que provoca el cambio de una guanina por una adenina que modifica el aminoácido valina por isoleucina en el primer dominio transmembrana del receptor y por ello fue designado CCR2-V64I (80). El alelo se traduce en una proteína funcional, sin modificación aparente de su actividad biológica. La frecuencia del alelo CCR2-64I es variable con el origen étnico pero, a diferencia del CCR5-D32, (25%), africanas (15%) e hispanas (17%) y es menos frecuente en individuos caucásicos (<10%). La amplia distribución en todos los grupos étnicos y su nivel de frecuencia sugiere un origen más antiguo que el CCR5-D32 (78).
El alelo CCR2-64I confiere una resistencia parcial a adquirir la infección por transmisión vertical (79)(80) pero no condicionaría el ingreso del HIV-1 por vía sexual o parenteral. Además se asocia con un retraso significativo de 2 años en la aparición de SIDA en los sujetos infectados tanto en la población adulta como pediátrica (80).
La presencia de ambas mutaciones, CCR5-D32 y CCR2-64I, es muy poco frecuente ya que la existencia de un desequilibrio de ligamiento entre ambos polimorfismos impide la existencia de ambas mutaciones en un mismo locus y, en consecuencia, condiciona la combinación de los genotipos, por lo que no existen los dobles homocigotas. La combinación de genotipos CCR2-64I y CCR5-D32 se halló predominantemente en el grupo de pacientes infectados LTNP, donde se correlaciona con una mejor evolución clínica. Aún no es claro el mecanismo por el cual una mutación puntual en la región transmembrana del receptor puede producir una mejoría clínica. Un efecto indirecto a través del CCR5 podría ser el responsable; sin embargo, no se ha podido demostrar que el CCR2-64I afecte la expresión y/o funcionalidad del CCR5. Por la proximidad a la que se encuentran ambos genes en el cromosoma 3 (3p21), a sólo 10 kilobases de distancia, se especuló que el CCR2-64I podría estar ligado a un polimorfismo en la región regulatoria del CCR5 y ejercer su efecto al modificar la expresión de ARN mensajero y la expresión del CCR5 en la superficie. Hasta el presente tampoco se ha podido comprobar esta hipótesis.
Además de polimorfismos en las regiones codificantes de los receptores de quimioquinas, se han identificado varios sitios polimórficos en la región regulatoria del CCR5 que influyen en la patogénesis del HIV-1 (71)(82-85). Ya se describieron 7 polimorfismos y varios presentan un fuerte ligamiento genético como el 29G con el D32, el 927T con el CCR2-64I y el 303A con el 627T. De acuerdo con los estudios de ligamiento se han definido 9 haplotipos humanos (HH) del CCR5 filogenéticamente relacionados de A-G (Fig. 4). Estudios de evolución realizados en primates permitieron identificar la secuencia más conservada y definir el haplotipo ancestral que corresponde al HHA. Los haplotipos restantes emergen por un proceso anidado (nest-ed) donde se producen polimorfismos de nucleótidos simples que se conservan a lo largo de la evolución (85). Según esta clasificación, los polimorfismos CCR5-D32 y CCR2-64I pertenecen a los haplotipos G2 y F2, respectivamente.
La distribución de los diferentes haplotipos varía de acuerdo con el origen étnico-geográfico de la población. El HHD se observó casi exclusivamente en poblaciones africanas, mientras que el HHC y HHE son más frecuentes en individuos caucásicos e hispanos (84)(86).
Debido a la condición bialélica de los genes, al analizar la distribución de las 81 combinaciones posibles de pares de HH del CCR5, el número de combinaciones observado fue limitado, con una frecuencia que difiere notoriamente de acuerdo con la etnia (86). En la población caucásica las combinaciones más frecuentes son el HHE/HHC (25%), HHC/HHC (11%), HHE/HHE (10%), representando aproximadamente 40% de todas las combinaciones de pares de haplotipos observados. Por el contrario, en la población africana ningún par de haplotipos supera el 10%.
En cuanto a la susceptibilidad a la infección por HIV-1, estudios en niños demuestran que el HHE y HHD facilitan la transmisión vertical del virus, mientras que HHC y HHF2 disminuyen la susceptibilidad. Tanto en niños como en adultos infectados, el alelo HHE acelera significativamente la progresión a SIDA y muerte. El efecto de los HHF2 y HHG2 fue previamente discutido como CCR5-D32 y CCR2-64I. La distribución de las combinaciones de haplotipos también difiere de acuerdo con el origen de la población: en individuos caucásicos los pares más frecuentes son HHE/HHC (25%), HHC/HHC (11%) y HHE/HHE (10%); por el contrario, en la población africana no predomina ninguna combinación. En adultos infectados de origen caucásico, el alelo HHC es el que se relaciona con una mejor evolución clínica combinado con HHG2 o HHE. En cambio, el genotipo homocigota HHE/HHE acelera la progresión a SIDA. Por el contrario, en poblaciones afroamericanas, el alelo HHC acelera la evolución a SIDA y los alelos HHA y HHF2 son los que la retrasan (86).

Figura 4. Árbol filogenético y haplotipos humanos (HH) del CCR5. Haplotipo ancestral HHA.
Participación de las quimioquinas y susceptibilidad a la infección por HIV-1
Además de los polimorfismos en los receptores de quimioquinas, variantes en las mismas pueden influir en la infección por HIV. Uno de ellos es el polimorfismo en el gen del SDF-1 denominado SDF1-3'A (guanina por adenina). La homocigosis para el alelo SDF1-3'A se asocia con una protección en la progresión a SIDA y muerte (87). Sin embargo, el papel protector es discutido (81)(84). Otro polimorfismo identificado en la CC-quimioquina RANTES, el RANTES-28G, aumenta el nivel de la expresión de la quimioquina y se asoció con retraso en la progresión a SIDA (87).
Recientemente se demostró que una isoforma no alélica de MIP-1a denominada MIP-1aP o CCL3L1 tiene una potente actividad supresora anti-HIV-1 y su gen puede presentar un número de copias variable (duplicaciones génicas) que puede ser de 0 a más de 10 copias por genoma diploide (89). Dosis génicas mayores se correlacionan con mayores niveles de la quimioquina en plasma. Además, se observó que para cada grupo étnico existe un número promedio de copias de CCL3L1 (90). Una dosis génica mayor que el promedio de la población se asocia con un fuerte efecto protector en la transmisión del HIV-1 y en el progreso a SIDA (90). Por ejemplo, en poblaciones de origen caucásico, el número promedio de copias es 2, mientras que en poblaciones africanas es 4; en ambos casos, en individuos con dosis génicas mayores que el promedio de la población se observa un fuerte efecto protector contra el HIV-1.
Marcadores genéticos en la infección pediátrica por HIV-1 en Argentina:
experiencia en el Hospital "J. P. Garrahan"
Poco después de la identificación de las variantes en los genes CCR5 y CCR2, en 1996, en el Hospital de Pediatría "J. P. Garrahan" se iniciaron los estudios genéticos en los co-receptores para evaluar su influencia en la transmisión vertical del HIV-1 y en la evolución a SIDA infantil. Inicialmente, se estudió la frecuencia de los alelos CCR5-D32 y CCR2-64I en población argentina a través de muestras obtenidas de donantes de sangre que concurrieron al Hospital Garrahan. Se demostró que los polimorfismos CCR5-D32 y CCR2-64I están presentes en esta población con frecuencias de 5% para el CCR5-D32 y 13% para el CCR2-64I. Una vez confirmada la presencia de los polimorfismos en esta población, se investigó la influencia de estas variantes en la infección infantil. La infección perinatal por el HIV-1 constituye un modelo valioso para estudiar determinantes genéticos por sus propias características. El HIV-1 es naturalmente transmitido al 13-48% de los niños nacidos de madres infectadas, por ello el riesgo de transmisión vertical es muy alto comparado con el riesgo de transmisión sexual (0,1-1%) (91).
Además, los niños nacidos de madres infectadas que no se infectaron son un grupo control ideal para comparar con el grupo infectado con el HIV-1, ya que a pesar del alto y comparable riesgo de exposición no adquirieron la infección. Otra característica importante es que en la infección por vía vertical es posible estimar en forma relativamente precisa el tiempo de transmisión. Además, la evolución clínica de la infección en los niños está bien caracterizada: aproximadamente 20% de los niños desarrollan rápidamente SIDA y mueren entre los 2-4 años de vida, mientras que la mayoría desarrolla la enfermedad en forma lenta, con una media de sobrevida de 8 años (92).
Los estudios realizados sobre un total de 882 niños nacidos de madres infectadas con el HIV-1, demuestran que el genotipo CCR5+/D32 no afecta la susceptibilidad a adquirir la infección por transmisión vertical, ni modifica la progresión de la enfermedad a diferencia de lo observado en adultos. Por el contrario, se demostró un efecto protector del CCR2-64I en la transmisión vertical del HIV-1 (70)(79) y en desarrollo de SIDA, el cual se vio retrasado en más de 3 años en niños portadores del alelo. A fin de comprender el efecto protector del CCR2-64I y profundizar los estudios relacionados con los genotipos del CCR5 y la infección por el HIV-1, se estudiaron 6 polimorfismos presentes en la región regulatoria del CCR5 y se analizaron según la clasificación de haplotipos. Estudiando la presencia de los distintos haplogrupos del CCR5 en la población argentina y la cohorte pediátrica expuesta al HIV-1, se halló que los haplogrupos más frecuentes son el HHC, HHE y HHF2 (85).
Analizando la distribución de los haplotipos del CCR5 presentes en el grupo de niños infectados y no infectados por el HIV-1, se identificó un conjunto de haplotipos del CCR5 asociados con diferentes grados de susceptibilidad a la transmisión vertical y en la progresión de la enfermedad, existiendo además una concordancia entre los haplotipos que afectan la transmisión viral y la progresión clínica (85). Estudiando la combinación de haplotipos, se observó que los pares de haplotipos HHE/HHC, HHE/HHE y HHE/HHG2 facilitan la transmisión vertical del HIV-1 y, por el contrario los pares HHC/HHG2 y HHC/HHC tienden a asociarse con una menor susceptibilidad a la infección. La mayor susceptibilidad se observó con el par HHE/HHG2 mientras que cuando el HHE se asocia con el HHF2 (alelo protector) se anula el efecto negativo. Por el contrario, cuando el HHC se combina con el HHG2 confiere el máximo efecto protector. Esto indica que el fenotipo asociado con un determinado genotipo depende de la interacción alelo-alelo, o sea que un genotipo puede asociarse con protección o no, según el alelo acompañante. En general, los pares de haplotipos del CCR5 que facilitan o dificultan la transmisión vertical concuerdan respectivamente con un efecto de aceleración o retraso en la progresión a SIDA en los niños infectados. Los tres pares de haplotipos con el alelo HHE que favorecen la transmisión también aceleraron la progresión de la enfermedad en los niños infectados.
En general se acepta que la posesión del alelo CCR5-D32 (haplotipo HHG2) está asociado con protección en el desarrollo de SIDA. Sin embargo, en la cohorte pediátrica estudiada no se observó tal protección. El estudio de los haplotipos del CCR5 permitió demostrar que el HHG2 puede asociarse con un aumento o reducción de la susceptibilidad a la infección dependiendo del alelo acompañante. Cuando el HHG2 se combina con el HHE se observó una mayor susceptibilidad y por el contrario cuando el HHG2 está acompañado por el HHC se observó una resistencia parcial a la transmisión viral. También se observó que el efecto fenotípico del alelo HHC o HHE en homocigosis (a pesar de ser opuestos) es menos marcado que en heterocigosis con el HHG2 (CCR5-D32), indicando que el alelo HHG2 fortalece el fenotipo del alelo acompañante. Estas observaciones reflejan que el efecto fenotípico asociado con un determinado genotipo del CCR5 es dependiente de las interacciones alelo-alelo y es la resultante de la suma de los efectos de los dos alelos. Este efecto también se observa en la progresión a SIDA, cuando el HHG2 está acompañado por el HHE los niños HIV-1 infectados acelerarían su progresión a SIDA. Por el contrario, cuando lo acompaña el HHC podría mejorar la evolución clínica retrasando la aparición de enfermedad.
Las poblaciones que son consideradas caucásicas presentan una gran heterogeneidad genética, por lo tanto la prevalencia de los haplotipos más frecuentes como el HHC, HHE y HHG2 pueden variar considerablemente entre los distintos grupos y por ello, la prevalencia de los pares HHC/HHG2 y HHE/HHG2 pueden ser muy diferentes en las distintas cohortes. Esto permite explicar por qué en los estudios iniciales restringidos a la heterocigosis CCR5-D32 no se encontró protección en la infección perinatal, ya que el 50% de los niños con el alelo HHG2 también poseían un alelo HHE. El estudio de la prevalencia de los distintos haplotipos del CCR5 en otras poblaciones posiblemente pueda explicar los resultados discordantes observados con respecto al genotipo heterocigota CCR5-D32 observado en la transmisión sexual en adultos de origen caucásico (59)(68)(69).
La estratificación genética y étnica de los individuos en las cohortes de estudio es importante para comprender mejor cómo las variaciones genéticas en el CCR5 influyen en las respuestas biológicas al HIV-1. También es importante para interpretar los resultados de estudios que combinan datos de varias cohortes, (como ser los meta-análisis), ya que los grupos pueden ser genéticamente muy heterogéneos y los efectos pueden enmascararse. Comparando estas observaciones con los resultados obtenidos en cohortes adultas, los distintos efectos de los alelos CCR5 en la evolución de la enfermedad podrían deberse a diferencias en la prevalencia de los diferentes alelos en las diferentes etnias.
Habiendo demostrado que variaciones genéticas en el CCR5 modifican la patogenia del HIV-1, la propuesta fue determinar si existe una relación inversa entre la cantidad de quimioquinas ligandos del CCR5 producido in vivo y el riesgo de infección por HIV-1. Para ello se estudió el MIP1-aP (CCL3L1) que es la quimioquina con mayor afinidad por el CCR5 y con fuerte actividad supresora anti-HIV, además su gen puede presentar duplicaciones segmentales por lo que el número de copias es variable. El promedio de copias de CCL3L1 hallado en la población argentina fue de 2, igual al hallado en americanos de origen europeo (90). En la cohorte de niños expuestos al HIV-1 se observó que los niños que portaban sólo 1 copia del gen presentaron mayor riesgo de adquirir la infección viral (70%) comparado con los niños que portaban 2 copias. Por el contrario, en los niños con más de 2 copias del gen CCL3L1 el riesgo disminuyó a medida que aumentaba el número de copias (Fig. 5) demostrando un rol protector.
Finalmente, se analizaron los resultados de variaciones cualitativas en CCR5/CCR2 con variaciones cuantitativas en CCL3L1. Para ello, se definieron grupos de riesgo genéticos (GRG) dicotomizando ambas variables en CCR5 detrimental y no detrimental (con o sin HHE) y CCL3L1 bajo o alto (número de copias menor o mayor a 2). Los resultados se muestran en la Tabla I. La combinación CCR5 no detrimental y CCL3L1 alto fue considerada el grupo de referencia y los riesgos se analizaron en relación a este grupo. A medida que se incorporaba una variable deletérea, se observó un aumento en el riesgo de transmisión, llegando a un riesgo 4 veces mayor con la combinación más perjudicial: CCR5 detrimental-CCL3L1 bajo (Tabla I).
Estos estudios demuestran que las variaciones genéticas en los co-receptores del HIV-1 desempeñan un rol relevante en la patogenia de la infección por el HIV-1, siendo marcadores genéticos pronósticos de infección y evolución de la enfermedad.
Tabla I. Riesgo de adquirir HIV-1 asociado con variaciones en CCL3L1 y/o CCR5.
CCL3L1alto : dosis génica mayor de 2; CCL3L1bajo: dosis génica menor o igual a 2; CCR5no-det: sin haplotipo HHE; CCR5det: con haplotipo HHE.


Figura 5. Dosis génica del CCL3L1 e infección por HIV-1. a) En gris claro se muestran los pacientes HIV-1 infectados y en gris más oscuro los niños expuestos al HIV-1 por transmisión vertical. b) Riesgo de adquirir HIV relativo a la mediana de la población/switch point. El número de copias promedio en la población argentina es de dos. OR= Odds Ratio, CI= intervalo de confianza 95%.
Conclusiones
Las variaciones genéticas en los receptores de quimioquinas -co-receptores del HIV-1- y en las quimioquinas juegan un papel relevante condicionando la infección por el HIV-1. Por ello, conocer el genotipo celular de las quimioquinas y sus receptores en cada población sería importante como marcadores pronósticos de susceptibilidad y evolución de la infección por el HIV-1 pero también para el diseño de vacunas y terapéuticas antirretrovirales más eficientes de acuerdo con las características genéticas de cada grupo.
Es importante destacar que no sólo las variaciones genéticas cualitativas (polimorfismos, mutaciones) sino también las cuantitativas (dosis génica) influyen en la susceptibilidad a la infección por HIV-1 y pueden constituir la base genética de la variabilidad de las respuestas a enfermedades infecciosas. El escenario con baja expresión del co-receptor CCR5 y alta producción de CC-quimioquinas es el más favorable para evitar la infección por las cepas de HIV-1 R5, que son las variantes involucradas en la mayoría de las transmisiones.
Correspondencia
DRA. ANDREA M. M. MANGANO
Laboratorio de Biología Celular y Retrovirus
Hospital de Pediatría "Prof. Dr. Juan P. Garrahan"
Combate de los Pozos 1881
1245 BUENOS AIRES, Argentina
Tel.: (54) (011) 4308-1998, 4308-4300 interno 1562
Fax: (54) (011) 4308-5325
E-mail: ammangano@retrovirus.org.ar
amangano@garrahan.gov.ar
Referencias bibliográficas
1. Centres for Disease Control. Update on acquired immunodeficiency syndrome (AIDS). Morbid Mortal Weekly Rep 1982; 31: 507-14.
2. Centres for Disease Control. Pneumocystis pneumonia. Morbid Mortal Weekly Rep 1981; 30: 250-2.
3. Centres for Disease Control. Kaposi's sarcoma and Pneumocystis pneumonia among homosexual men. Morbid Mortal Weekly Rep 1982; 30: 305-8.
4. Masur H, Michelis MA, Greene JB, Onorato I, van de Stouwe RA, Holzman RS, et al. An outbreak of community acquired Pneumocystis carinii pneumonia: initial manifestation of cellular immune dysfunction. New Engl J Med 1981; 305: 1.431-8.
5. Levy JA, Hoffman AD, Kramer SM, Landis JA, Shimabukuro JM, Oshiro LS. Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science 1984; 225: 840-2.
6. Centres for Disease Control. Possible transfusion-associated acquired immunodeficiency syndrome (AIDS). Morbid Mortal Weekly Rep 1982; 31: 652-4.
7. Centres for Disease Control. Unexplained immunodeficiency and opportunistic infections in infants. Morbid Mortal Weekly Rep 1982; 31: 665-7.
8. Centres for Disease Control. Immunodeficiency among female sexual partners of males with acquired immunodeficiency syndrome in New York. Morbid Mortal Weekly Rep 1982; 31: 697-8.
9. Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Charamet S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983; 220: 868-70.
10. Gallo RC, Sarin PS, Gelman EP, Robert-Guroff M, Richrdson E, Kalyanaraman VS, et al. Isolation of human T cell leukemia virus in acquired immunodeficiency syndrome (AIDS). Science 1983; 220: 865-7.
11. Gallo RC, Zalahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haayneess BF, et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984; 224: 500-2.
12. Guo HG, Cherman JC, Waters D, Hall L, Louie A, Gallo RC, et al. Sequence analysis of original HIV-1. Nature 1991; 349: 745-6.
13. Clavel F, Guétard D, Brun Vézinet F, Charmaret S, Ray MA, Santos Ferreira MO, et al. Isolation of a new human retrovirus form West African patients with AIDS. Science 1986; 233: 343-6.
14. Gao F, Bailes E, Robertson DL, Chen Y, Rodenburg CM, Michael SF, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 1999; 397: 436-41.
15. Weiss RA, Wrangham RW. From pan to pandemic. Nature 1999; 397: 385-6.
16. Chen Z, Luckay A, Sodora DL, Telfer P, Reed P, Geetie A, et al. Human immunodeficiency virus type 2 (HIV 2) seroprevalence and characterization of a distinct HIV-2 genetic subtype from the natural range of simian immunodeficiency virus-infected sooty mangabeys. J Virol 1997; 70: 3.953-60.
17. Myers G, Korber B, Foley B, Jeang KT, Mellors JW, Wain Hobson S. Eds. Human Retroviruses and AIDS: a compilation and analysis of nucleic acid and amino acid sequences. Los Alamos. New Mexico: Los Alamos National Library; 1996.
18. Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature 1998; 391: 594-7.
19. A global view of HIV infection: 2006 Global Report prevalence map. Report on the global AIDS epidemic, UNAIDS. [Available on: www.unaids.org] Accessed May 2006.
20. Pantaleo G, Graziosi C, Fauci AS. The immunopathogenesis of human immunodeficiency virus infection. N Eng J Med 1993; 328: 327-35.
21. Koup RA, Safrit JT, Cao Y. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol 1994; 68: 4.650-5.
22. Prince AM, Reesnick H, Pascual D. Prevention of HIV infection by passive immunizations with HIV immunoglobulin. AIDS Res Hum Retrovirus 1991: 7: 971-4.
23. Bukowski JF, Warner JF, Dennert G, Welsh RM. Adoptive transfer studies demonstrating the antiviral effect of natural killer cells in vivo. J Exp Med 1985; 161: 40-52.
24. Shearer WT, Kalish LA, Zimmerman PA. CCR5 HIV-1 vertical transmission. Women and infants transmission group. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 17: 180-1.
25. Groux H, Torpier G, Monte D, Mouton Y, Capron A, Ameisen JC. Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. J Exp Med 1992; 175: 331-40.
26. Amadori A, De Silvestro G, Zamarchi R, Veronese ML, Mazza MR, Schiavo G, et al. CD4 epitope masking by CD4+ cell function down-regulation in AIDS patients. J Immunol 1992; 148: 2.709-16.
27. Clerici M, Wynn TA, Berzofky JA, Blatt SP, Hendrix CW, Sher A, et al. Role of interleukine 10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest 1994; 93: 768-75.
28. Shearer GM, Clerici M. Early T-helper cell defects in HIV infection. AIDS 1991; 5: 245-53.
29. Pantaleo G, Fauci AS. Tracking HIV during disease progression. Curr Opin Immunol 1994; 6: 600-4.
30. Safrit JT, Koup RA. The immunology of primary HIV infection: which immune responses control HIV replication? Curr Op Immunol 1995; 4: 456-61.
31. Mellors JW, Rinaldo CR Jr, Gupta P, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 1996; 272: 1.167-70.
32. Cheng-Mayer C, Seto D, Tateno M, Levy JA. Biological features of HIV that correlates with virulence in the host. Science 1998; 240: 80-2.
33. Fenÿo E, Morfeldt-Mason L, Chiodi F, Lind B, VongGerfelt A, Albert J, et al. Distintive replicative and cythopatic characteristics of human immunodeficiency virus isolates. J Virol 1998; 62: 4.414-9.
34. Fauci A. Host factors and the pathogenesis of HIV-1 induced disease. Nature 1996; 384: 529-34.
35. Tersmette M, Gruters RA, DeWolf F, DeGoude REY, Lange JMA, Schellekens PTA, et al. Evidence for a role of a virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency virus syndrome: studies on sequential isolates. J Virol 1989; 63: 2.118-25.
36. Shuitemaker H, Kootstra NA, De Goede Rey, de Wolf F, Miedema F, Tersmette M. Monocytotropic human immunodeficiency virus 1 (HIV-1) variants detectable in all stages of HIV infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J Virol 1991; 65: 356-63.
37. Shuitemaker H. Macrophage-tropic HIV-1 variants: initiators of infection and AIDS pathogenesis? J Leuk Biol 1994; 56: 218-24.
38. Koot M, Keet IPH, Vos AHV, de Goede REY, Roos M, Coutubho RA, et al. Prognostic value of HIV-1 syncitium inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med 1993; 118: 681-8.
39. Masciotra S, Livelava B, Belloso W, Clave L, Tanuri A, Ramos AC, et al. Evidence of a high frequency of HIV-1 subtype F infections in a heterosexual population in Buenos Aires, Argentina. AIDS Res Hum Retroviruses 2000; 10: 1.007-14.
40. Neilson J, John G, Can JK, Lewis P, Kweiss JK, Jacbson S, et al. Subtypes of human immunodeficiency virus type 1 and disease stage among women in Nairobi, Kenya. J Virol 1999; 73: 4.393-403.
41. Alkhatib G, Broder CC, Berger EA. Cell type-specific cofactors determine human immunodeficiency virus type 1 tropism for T-cell lines versus primary macrophages. J Virol 1996; 70: 5.487-94.
42. Berson JF, Long D, Doranz BJ, Jirik FR, Doms RW. A seven transmembrane domain receptor involved in fusion and entry of T-cell tropic human immunodeficiency virus type-1 strains. J Virol 1996; 70: 6.288-95.
43. Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven transmembrane G protein coupled receptor. Science 1996; 272: 872-7.
44. Paxton WA, Martin SR, Tse D, O'Brien TR, Skurnick J, VanDevanter NL, et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposures. Nature Med 1996; 2: 412-7.
45. Cochi F, DeVico AL, Garzimo-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1a and MIP-1b as the major HIV-suppressive factor produced by CD8+ T cells. Science 1995; 270: 1.811-5.
46. Paxton WA, Liu R, Kamg S, Wu L, Gingeras TR, Landau NR, et al. Reduced HIV-1 infectability of CD4+ lymphocytes from exposed-uninfected individuals: association with low expression of CCR5 and high production of b-chemokines. Virology 1998; 244: 66-73.
47. Deng HK, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhaart M, et al. Identification of a major coreceptor for primary isolates of HIV-1. Nature 1996; 381: 661-6.
48. Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptors CC-CKR5. Nature 1996; 381: 667-73.
49. Bleul CC, Farzan M, Ca H, Paolini C, Clark-Lewis I, Sodoski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 1996; 382: 829-32.
50. Oberlin E, Amara A, Bachelerie ]F, Bessia C Virelizier JL, Arenzana-Seisdedos F, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 1996; 382: 833-5.
51. Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, et al. The b-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996; 86: 1.135-48.
52. Doranz BJ, Rucker J, Yi J, Smyth RJ, Samson M, Peiper SC, et al. A dual-tropic, primary HIV-1 isolate that uses fusin and the b-chemokine receptors CKR-5, CKR3 abd CKR-2b as fusion cofactors. Cell 1996; 86: 1.149-59.
53. Murphy PM. Chemokine receptors: structure, function and role in microbial pathogenesis. Cytokine Growth Fact Rev 1996; 12: 593-633.
54. Deng HK, Unutmaz D, Kewalramani VN, Littman DR. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 1997; 388: 296-300.
55. Llao F, Alkhatib G, Peden KWC, Sharma G, Berger EA, Farber JM. STRL-33, a novel chemokine receptor-like protein, functions as a fusion cofactor for both macrophage-tropic and T-cell line-tropic HIV-1. J Exp Med 1997; 185: 2015-23.
56. Berger EA, Doms RW, Fenyo E-M. A new classification for HIV-1. Nature 1998; 391: 621-8.
57. Moore JP, Trkola A, Dragic T. Co-receptors for HIV-1 entry. Curr Opin Immunol 1997; 9: 551-62.
58. Liu R, Paxton WA, Choe S, Ceradini D, Marin SR, Horuk R, et al. Homozygous defect in HIV-1 co-receptor accounts for resistance in some multiply-exposed individuals to HIV-1 infection. Cell 1996; 86: 367-77.
59. Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber C-M, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996; 382: 722-5.
60. Huang Y, Paxton WA, Wolinsky SM, Neuman AU, Zhang L, He T, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nature Med 1996; 2: 1.240-3.
61. Martinson JJ, Hong L, Karanicolas R, Moore JP, Kostrikis LG. Global distribution of the CCR2-64I/CCR5-59653T HIV-1 disease-protective haplotype. AIDS 2000; 14: 483-9.
62. Biti R, French R, Young J, Bennett B, Stewart G. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med 1997; 3: 252-3.
63. O'Brien TR, Winkler C, Dean M, Nelson JA, Carr E. HIV-1 infection in an individual homozygous for the CCR5 deletion mutant. Lancet 1997; 349: 1.219.
64. Theodorou I, Meyer L, Magierowska M, Katlana C, Rouzioux C. HIV-1 infection in an individuañ homozygous for CCR5 delta-32. Lancet 1997; 349: 1.219.
65. Ballota C, Bagnarelli P, Violin M, Ridolfo AL, Zhou D, Berlusconi A, et al. Homozygous D32 deletion of the CCR5 chemokine receptor gene in an HIV-1 infected patient. AIDS 1997; 11: F67-F71.
66. Heiken, H., Becker, S., Bastisch, I., Schmidt, R. HIV-1 infection in a heterosexual man homozygous for CCR5 delta 32. AIDS 1999; 13: 529-30.
67. Kuipers H, Workman C, Dyer W, Geczy A, Sullivan J, Oelsichs P. An HIV-1 infected individual homozygous for the CCR5 delta 32 allele and the SDF-1 3'A allele. AIDS 2000; 13: 433-4.
68. Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikments R, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996; 273: 1.856-62.
69. Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, Birx DL, et al. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nat Med 1997; 3: 338-40.
70. Mangano A, Prada F, Roldán A, Picchio G, Bologna R, Sen L. Distribution of CCR-5 D32 allele in Argentinian children at risk of HIV-1 infection: its role in vertical transmission. AIDS 1998; 12: 109-10.
71. Misrahi M, Teglas JP, N'Go N, Burgard M, Mayaux M-J, Rouzioux C, et al. CCR5 chemokine receptor variant in HIV-1 mother-to-child transmission and disease progression in children. JAMA 1998; 279: 277-80.
72. Ioannidis JP, O'Brien TR, Rosenberg PS, Contopoulos-Ioannidis DG, Goedert JJ. Genetic effects on HIV disease progression. Nat Med 1998; 4: 536.
73. Martinson JJ, Chapman NH, Rees DC, Liu YT, Clegg J. Global distribution of the CCR5 gene 32-base pair deletion. Nature Genet 1997; 16: 100-3.
74. Mangano A, Theiler G, Sala L, Capucchio M, Fainboim L, Sen L. Distribution of CCR5-Delta 32 and CCR2-64I alleles in an Argentine amerindian population. Tissue Antigens 2001 ; 58: 99-102.
75. Rocco CA, Mangano A, del Pozo A, Sen L. Distribution of CCR5-CCR2 haplotypes in an Argentinian population. AIDS Res Hum Retroviruses 2003;19: 943-5.
76. Libert F, Cochaux P, Beckman G, Samson M, Akesenova M, Cao A, et al. The Dccr5 mutation conferring protection against HIV-1 in Caucasians populations has a single and recent origin in Northeastern Europe. Hum Mol Genet 1998; 7: 399-406.
77. Sthephens JC, Reich DE, Golstein DB, Doo Shin H, Smith M, Carrington M, et al. Dating the origin of the CCR5-D32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 1998; 62: 1.507-15.
78. Martinson JJ, Hong L, Karanicolas R, Moore JP, Kostrikis LG. Global distribution of the CCR2-64I/CCR5-59653T HIV-1 disease-protective haplotype. AIDS 2000; 14: 483-9.
79. Mangano A, Kopka J, Batalla M, Bologna R, Sen L. Protective effect of CCR2-64I and not of CCR5-D32 and SDF1-3'A in Pediatric HIV-1 Infection. JAIDS 2000, 23: 52-7.
80. Smith MW, Dean M, Carrington M, Winkler C, Huttley GA, Lomb DA, et al. Constransting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Science 1997; 277: 959-64.
81. Mummidi S, Ahuja SS, Gonzalez E, Andreson SA, Santiago EN, et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat Med 1998; 4: 786-93.
82. Kostrikis FG, Huang Y, Moore JP, Wolinsky SM, Zhang L, Guo Y. A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promotor mutation. Nat Med 1998; 4: 350-3.
83. Kostrikis L, Neumann A, Thomson B, Korber BT, McHardy P, Karanicolas R, et al. A polymorphism in the regulatory region of the CC-chemokine receptor 5 gene influences perinatal transmission of human immunodeficiency virus type 1 to African-American children. J Virol 1999; 73: 10.264-71.
84. Mangano A, Kopka J, Batalla M, Bologna R, Sen L. Protective effect of CCR2-64I and not of CCR5-D32 and SDF1-3'A in pediatric HIV-1 infection. JAIDS 2000; 23: 52-7.
85. Mangano A, Gonzalez E, Dhanda R, Catano G, Bamshad M, Bock A, et al. Concordance between the CC chemokine receptor 5 genetic determinants that alter risks of transmission and disease progression in children exposed perinatally to human immunodeficiency virus. JID 2001;183: 1574-85.
86. Gonzalez E, Bamshad M, Sato N, Mummidi S, Dhanda R, Catano G, et al. Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc Natl Acad Sci USA 1999; 96: 12.004-9.
87. Winkler C, Modi W, Smith MW, Nelson GW, Wu X, Carrington M, et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. Science 1998, 279: 389-93.
88. Liu H, Chao D, Nakayama EE, Taguchi H, Goto M, Xin X, et al. Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proc Natl Acad Sci USA 1999. 13: 4.581-5.
89. Nibbs RJ, Yang J, Landau NR, Mao JH, Graham GJ. LD78beta, a non-allelic variant of human MIP-1alpha (LD78alpha), has enhanced receptor interactions and potent HIV suppressive activity. J Biol Chem 1999; 274: 17.478-83.
90. Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 2005; 307: 1.434-40.
91. Royce RA, Sena A, Cates W Jr, Cohen MS. Sexual transmission of HIV. New Engl J Med 1997; 336: 1.072-8.
92. Wilfert CM, Wilson C, Luzuriaga K. Pathogenesis of pediatric human immunodeficiency virus type 1 infection. J Infect Dis 1994; 170: 286-92. [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ] [ Links ]
Aceptado para su publicación el 28 de septiembre de 2007

