










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.45 no.1 La Plata ene./mar. 2011
TEMA DE INTERÉS
Guías de práctica de laboratorio clínico
Uso de los marcadores tumorales en el cáncer de hígado, de vejiga, cervical y gástrico
Capítulos 1 a 3
EDITADO POR
Catharine M. Sturgeon
Eleftherios P. Diamandis
Catharine M. Sturgeon
Department of Clinical Biochemistry, Royal Infirmary of Edinburgh, Edinburgh, Reino Unido
Michael J. Duffy
Department of Pathology and Laboratory Medicine, St Vincent's University Hospital and UCD School of Medicine and Medical Science, Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Dublín, Irlanda
Barry R. Hoffman
Department of Pathology and Laboratory Medicine, Mount Sinai Hospital, and Department of Laboratory Medicine and Pathobiology, University of Toronto, Ontario, Canadá
Rolf Lamerz
Department of Medicine, Klinikum of the University Munich, Grosshadern, Alemania
Herbert A. Fritsche
Department of Laboratory Medicine, The University of Texas M. D. Anderson Cancer Center, Houston, TX
Katja Gaarenstroom
Department of Gynecology, Leiden University Medical Center, Leiden, Holanda
Johannes M.G. Bonfrer
Department of Clinical Chemistry, Netherlands Cancer Institute, Amsterdam, Holanda
Thorsten Ecke
Department of Urology, Helios Hospital, Bad Saarow, Alemania
H. Barton Grossman
Department of Urology, The University of Texas M. D. Anderson Cancer Center, Houston, TX
Peter Hayes
Scottish Liver Transplant Unit, Department of Medicine, Royal Infirmary of Edinburgh, Edinburgh, Reino Unido
Ralf-Thorsten Hoffmann
Department of Clinical Radiology, LMU-Klinikum- Grosshadern, University of Munich, Alemania
Seth P. Lerner
Department of Urology, Baylor College of Medicine, Houston, TX
Florian Lohe
Department of Surgery, LMU-Klinikum-Grosshadern, University of Munich, Alemania
Johanna Louhimo
Department of Clinical Chemistry, Helsinki University Central Hospital, Finlandia
Ihor Sawczuk
Department of Urology, Hackensack University Medical Center, Hackensack, NJ
Kazuhisa Taketa
Clinical Trial Center, Brain Attack Center, Oota Memorial Hospital, Fukuyama, Japón
Eleftherios P. Diamandis
Department of Pathology and Laboratory Medicine, Mount Sinai Hospital, and Department of Laboratory Medicine and Pathobiology, University of Toronto, Ontario, Canadá
Este documento ha sido traducido con permiso de la National Academy of Clinical Biochemistry (NACB), Washington, DC. La NACB no se hace responsable de la exactitud de la traducción. Los puntos de vista presentados son los de los autores y no necesariamente los de la NACB.
Índice
Capítulo 1. Introducción
Capítulo 2. Marcadores tumorales en cáncer de hígado
Capítulo 3. Marcadores tumorales en cáncer de vejiga
Capítulo 4. Marcadores tumorales en cáncer cervical
Capítulo 5. Marcadores tumorales en cáncer gástrico
Referencias
Agradecimientos
Apéndice
Referencias del apéndice
Introducción
Este documento presenta a los bioquímicos, médicos y a otros miembros del laboratorio clínico la actualización más reciente de las Guías de Práctica del Laboratorio Clínico de la National Academy of Clinical Biochemistry (NACB) para uso de los marcadores tumorales en los cánceres de hígado, vejiga, cervical y gástrico. Estas guías tienen como objetivo fomentar un uso más apropiado de los marcadores tumorales por parte de los médicos en la atención primaria, en la atención hospitalaria, cirujanos, especialistas oncólogos y otros profesionales de la salud.
Las guías de práctica clínica son afirmaciones desarrolladas sistemáticamente que pretenden ayudar a los médicos y a los pacientes en sus tomas de decisiones con respecto a una atención de la salud apropiada en circunstancias clínicas específicas (1). Se publicó con anterioridad una explicación de los métodos utilizados al desarrollar estas guías (2) y se la incluyó como un Apéndice de este documento. Tal como podría esperarse, muchas de las recomendaciones de la NACB son similares a aquellas que ofrecen otros grupos, tal como se puede apreciar en las comparaciones tabulares que se presentan para cada malignidad (2).
Para elaborar estas pautas, se revisó la literatura relevante relacionada con el uso de los marcadores tumorales. Se prestó particular atención a las revisiones, entre ellas las pocas revisiones sistemáticas relevantes, y a las guías emitidas por los paneles de expertos. En lo posible, las recomendaciones consensuadas del panel de la NACB informadas aquí se basaron en evidencias disponibles (es decir, se basaron en la evidencia). Anteriormente se publicaron recomendaciones de la NACB relativas a los requerimientos de calidad general para las mediciones de marcadores tumorales, entre las que se encuentra la tabulación de causas importantes de resultados falso-positivos que también deben ser tenidos en cuenta (por ejemplo, interferencia por anticuerpos heterofílicos,highdose hooking) (3).
Marcadores tumorales en cáncer de hígado
Antecedentes
El carcinoma hepatocelular (CHC) es la quinta causa más común de cáncer en hombres y la octava causa más común de cáncer en mujeres en todo el mundo (4) (5). También es la tercera causa más común de muerte por cáncer (6), con un diagnóstico de 500.000 casos nuevos por año. La incidencia mundial ajustada por edad varía por área geográfica, con un incremento de 5,5/100.000 de la población en los Estados Unidos y Europa a 14,9/100.000 en Asia y África (7). La incidencia más alta que se ha observado en Europa durante la última década, probablemente refleje el creciente número de casos de infección por hepatitis C (8) (9) y cirrosis hepática (10), ambos fuertes factores predisponentes para el CHC (11).
En la mayoría de los lugares de Asia y África, la infección por el virus de la hepatitis B es más relevante (12), siendo la ingesta de aflatoxina B1 por alimentos contaminados un factor contribuyente adicional (13). En Occidente y en Japón, la infección por el virus de la hepatitis C es el factor de riesgo principal (7)(14-17), aunque los pacientes con cirrosis alcohólica o hemocromatosis también tienen un riesgo elevado (18). En esta parte del mundo, los pacientes mayores son más propensos a desarrollar el CHC que los más jóvenes (15) (16). Por otro lado, en los países en desarrollo, el CHC afecta más frecuentemente a los individuos más jóvenes que tienen hepatitis B crónica (19), mientras que los portadores tienen doble riesgo relativo de desarrollar la enfermedad. Los pacientes cirróticos tienen un riesgo más alto que los no cirróticos, con incidencias anuales del CHC de 2% a 6,6% (20) y 0,4% (21), respectivamente. En todo el mundo, 380 millones de individuos están infectados con hepatitis B y 170 millones con hepatitis C (22). Es posible realizar una vacunación preventiva contra la hepatitis B pero no contra la hepatitis C. Hay nuevas estrategias antivirales terapéuticas disponibles (por Ej. interferón alfa pegilado combinado con ribavirina u otros medicamentos como la lamivudina) para el tratamiento de la hepatitis B y C (23-25).
Los fundamentos detrás del screening para la detección del CHC mediante ultrasonido hepático regular y medición de marcadores tumorales en grupos de alto riesgo asintomáticos, son que el screening facilita la identificación temprana de tumores cuando todavía son potencialmente curables. En pacientes con cirrosis o hepatitis viral crónica controlados de esta manera, una concentración elevada de alfa fetoproteína sérica (AFP) puede ser la primera indicación de un tumor maligno, requiriendo un diagnóstico por imágenes adicional del hígado y mayor investigación (26). En un paciente asintomático, un nódulo sólido predominante que no es consistente con un hemangioma puede indicar CHC (27), mientras que las lesiones hipervasculares asociadas con AFP elevada (> 400 μg/L) son casi diagnósticas de cáncer. Idealmente, se deberían llevar a cabo pruebas controladas aleatorias para demostrar la eficacia del screening en términos de menor mortalidad por enfermedad y mayor supervivencia y costo-efectividad (28). Es improbable que se lleven a cabo dichas pruebas, porque ya está generalmente aceptado que implementar un control sistemático es beneficioso para ciertos pacientes cirróticos (29). Actualmente, en los países desarrollados, aproximadamente 30%-40% de los pacientes con CHC son diagnosticados con suficiente anticipación como para recibir tratamientos curativos.
Debido a que muchos pacientes con enfermedad temprana son asintomáticos (30) (31), el CHC suele diagnosticarse tarde, cuando generalmente ya es intratable (32). La sospecha de enfermedad puede surgir primero en pacientes con cirrosis hepática que desarrollan ascitis, encefalopatía o ictericia (33). Algunos pacientes inicialmente presentan dolor abdominal superior, pérdida de peso, saciedad temprana o masa palpable en el abdomen superior (31). Otros síntomas incluyen ictericia obstructiva, diarrea, dolor óseo, disnea, sangrado intraperitoneal, síndrome paraneoplástico [por ej., hipoglicemia (34), eritrocitosis (35), hipercalcemia (36) (37)], diarrea acuosa severa (37) o manifestaciones cutáneas (por ej. dermatomiositis) (38).
Las modalidades de diagnóstico por imágenes incluyen ultrasonido, tomografía computarizada (TC) e IRM (6)(39). El ultrasonido es ampliamente accesible, no invasivo y se usa generalmente en pacientes con CHC para evaluar el aporte sanguíneo a nivel hepático y la invasión vascular del tumor, así como también intraoperativamente para detectar pequeños nódulos tumorales. Aunque la TC de hígado se usa ocasionalmente para investigar las anormalidades identificadas en el ultrasonido, rara vez se la utiliza como el método de screening principal. Las pautas de la American Association for the Study of Liver Diseases (AASLD) indican específicamente que no hay datos que avalen el control con TC (40). La IRM brinda imágenes del hígado de alta resolución.
Los especímenes para la histopatología generalmente se obtienen por biopsia con asistencia de ultrasonido o TC. La biopsia implica el riesgo de que se expanda el tumor por el trayecto de la aguja (1%-2,7% total) (41) (42). La apariencia histológica del CHC varía de lesiones bien a mal diferenciadas de células gigantes tumorales anaplásticas de núcleos múltiples grandes, con necrosis central frecuente. Existe un continuo debate sobre la relevancia de clasificar la displasia en la predicción del CHC.
Excepto en Japón, rara vez se diagnostica a los pacientes con CHC en los primeros estadios del carcinoma maligno in situ (43), cuando las tasas de supervivencia a 5 años son de 89%-93% después de la resección y 71% después del tratamiento percutáneo (44). Los pacientes con CHC temprano tienen un nódulo tumoral < 5 cm o 2-3 nódulos < 3 cm cada uno. El pronóstico depende de la cantidad y tamaño del o los nódulos, de la función hepática al momento del diagnóstico, y de la elección de tratamiento (45) (46). La mayor heterogeneidad de la enfermedad vista en enfermedades más avanzadas complica la selección del tratamiento óptimo, que a la vez se refleja en la variación considerable de las tasas de supervivencia informadas en pruebas aleatorias controladas (por ej. 1 año, 10%-72%, 2 años 8%-50%) (47).
En los centros de derivación de los países occidentales se les brindan tratamientos curativos al 30%-40% de los pacientes con CHC y en Japón, al 60%-90% de los pacientes (6). La resección hepática es el tratamiento de elección en pacientes no cirróticos, con un un 70% de supervivencia a 5 años en pacientes cuidadosamente seleccionados. Se pueden alcanzar tasas de supervivencia altas similares mediante el transplante en pacientes cirróticos seleccionados adecuadamente (por ej. con 1 nódulo < 5 cm de diámetro o hasta 3 nódulos < 3 cm cada uno). Recientemente se ha revisado el manejo moderno del CHC (40) (48) (49).
Entre los tratamientos potenciales se encuentran la ablación percutánea, la quimioembolización y la quimioterapia. Los tratamientos percutáneos brindan las mejores opciones de tratamiento para el CHC temprano no extirpable, mientras que la destrucción de las células neoplásicas se alcanza mediante tratamientos químicos (alcohol, ácido acético) o físicos (radiofrecuencia, microondas, láser, crioablación) (50). La inyección percutánea de etanol se ha asociado con pocos eventos adversos, tasas de respuesta de hasta 90%-100% y tasas de supervivencia a 5 años de hasta 50% (51) en grupos de pacientes seleccionados. La ablación por radiofrecuencia o la inyección de etanol son muy efectivas en pacientes con un tumor < 3 cm. La ablación por radiofrecuencia también es efectiva, con respuestas objetivas comparables, siendo necesarias menos sesiones (52) y con mejores tasas de supervivencia a 5 años en pacientes con tumores más grandes (53) (54).
Los tratamientos paliativos en enfermedad avanzada incluyen quimioembolización arterial, con ventajas de supervivencia en candidatos bien seleccionados (47). Los agentes de embolización como Gelfoam administrado con agentes de quimioterapia selectivos (por ej. doxorubicina, mitomicina o cisplatina) mezclados con lipiodol (quimioembolización) pueden demorar la progresión del tumor y la invasión vascular en 15%-55% de los pacientes. Se están desarrollando terapias moleculares para el CHC (55) sobre la base de mejorar la comprensión y detección de la activación atípica de varias cascadas de señales involucradas en la transformación de las células hepáticas. En pruebas controladas por placebo, multicéntricas fase III, una de estas drogas nuevas -el inhibidor multiquinasa sorafenib- demostró ser modestamente eficaz en el tratamiento del CHC en estadio avanzado [Clasificación de Barcelona Clinic Liver Cancer (BCLC) estadios B y C; (55-57)].
De la discusión anterior se desprende que para un resultado favorable es conveniente la detección temprana del CHC, preferentemente cuando todavía es asintomático. El objetivo de este informe es presentar nuevas Guías de la NACB para el uso de marcadores tumorales tisulares y séricos en la detección temprana del CHC y su manejo. Para elaborar estas pautas se consultó la literatura relevante para el uso de marcadores tumorales en el CHC. Se prestó especial atención a las revisiones, incluyendo las revisiones sistemáticas de las pruebas aleatorias prospectivas que incluyeron el uso de marcadores y guías emitidas por paneles de expertos. Cuando fue posible, las recomendaciones consensuadas del panel de la NACB se basaron en evidencias disponibles (es decir, se basaron en pruebas). También se presenta un resumen de las guías sobre estos temas publicadas por otros paneles de expertos.
Marcadores disponibles para el CHC
En la Tabla I se enumeran los marcadores tumorales séricos y tisulares más ampliamente investigados para el CHC, junto con la fase de desarrollo de cada marcador y el nivel de evidencia (LOE, por sus sigla en inglés) de su uso clínico (58; nivel 1, evidencia de un estudio controlado prospectivo de alta potencia que fue específicamente diseñado para probar el marcador o evidencias de un metaanálisis, análisis comunes o descripción general de estudios nivel II o III; nivel II, evidencias de un estudio en el cual los datos del marcador están determinados en relación con una prueba terapéutica prospectiva que se realiza para probar la hipótesis terapéutica pero no específicamente diseñada para probar la utilidad del marcador; nivel III, evidencia de grandes estudios prospectivos; nivel IV, evidencia de pequeños estudios prospectivos; nivel V, evidencia de pequeños estudios piloto). De los marcadores enumerados, sólo la AFP se utiliza ampliamente en la práctica clínica.
Tabla I. Marcadores séricos y tisulares actualmente disponibles para el cáncer de hígado
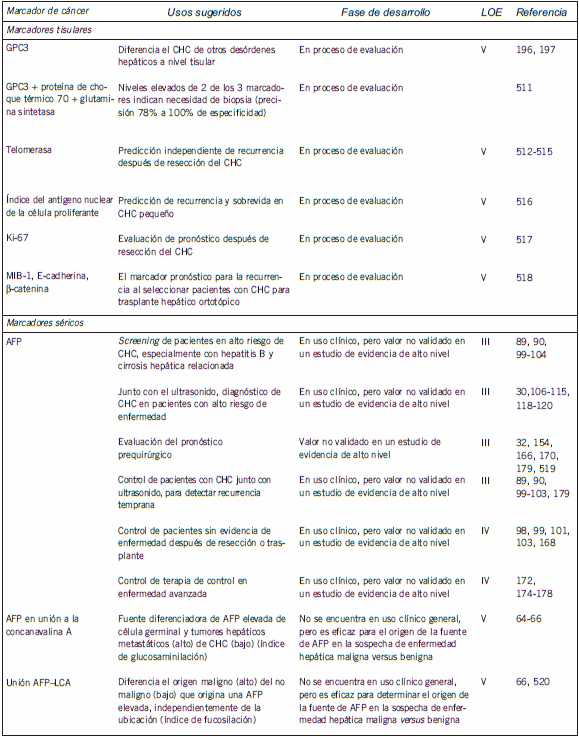
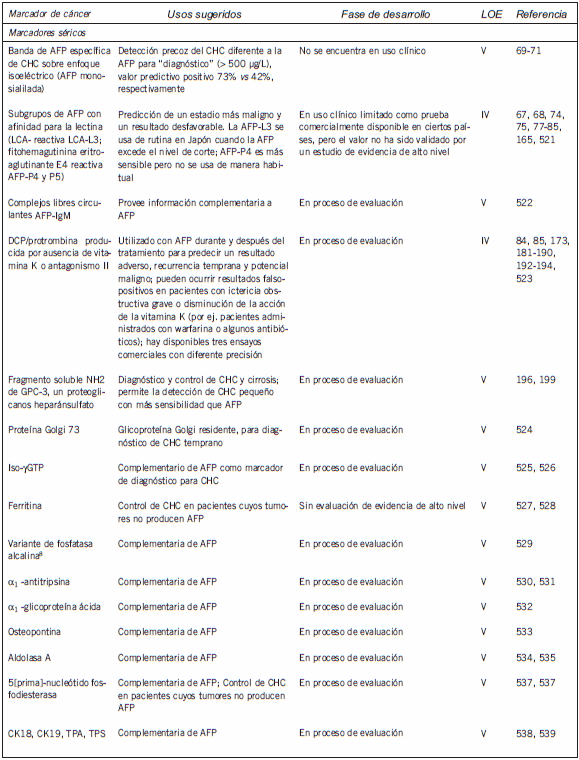
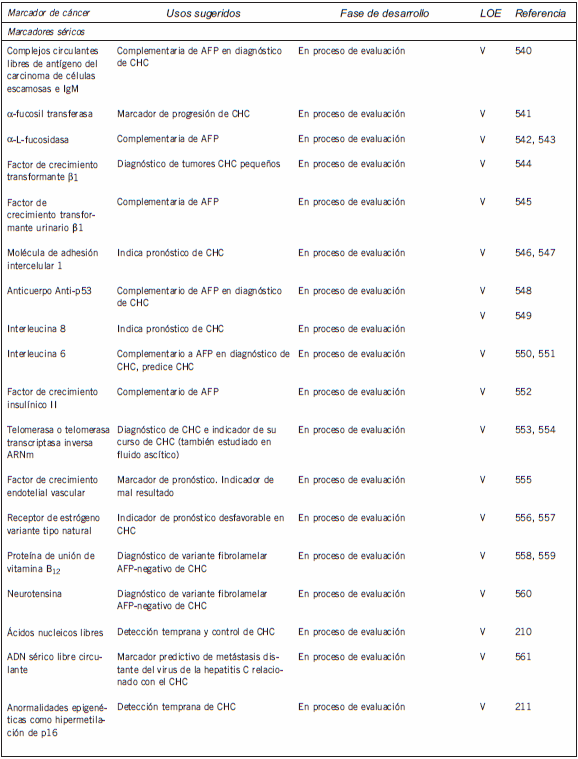
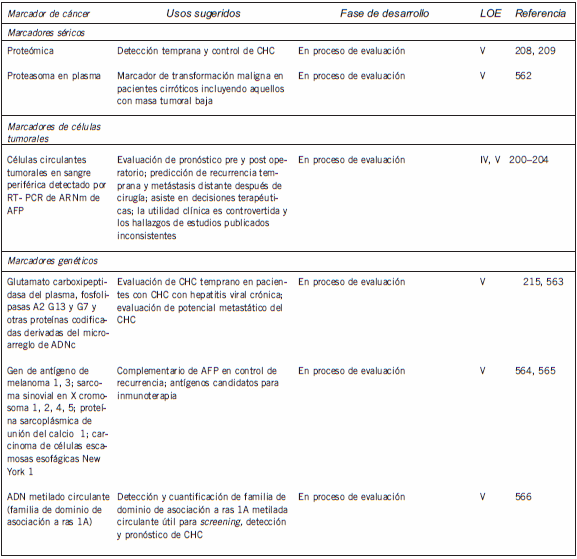
Marcadores tumorales en cáncer de hígado: recomendaciones de la NACB
En la Tabla II se presenta un resumen de las recomendaciones de las pautas representativas publicadas sobre el uso de la AFP en el CHC, que también sintetiza las guías actuales de la NACB para el uso de los marcadores en este cáncer. A continuación, se presenta una discusión más detallada de algunos de los marcadores enumerados en las Tabla I y II.
Tabla II. Recomendaciones para uso de la AFP en cáncer de hígado por diferentes grupos de expertos
Alfa-fetoproteína
La AFP es una glicoproteína de 70-kD que consiste en 591 aminoácidos y 4% de residuos de hidratos de carbono , codificados por un gen en el cromosoma 4q11-q13 [para reseñas, ver (50) (60)]. Durante la gestación, normalmente el hígado y el saco vitelino del feto producen la AFP, y está muy elevada en la circulación de los recién nacidos. Sin embargo, sus concentraciones disminuyen durante los siguientes 12 meses a 10-20 μg/L.
CONSIDERACIONES ANALÍTICAS
Métodos de ensayo, estandarización y valores de referencia
Actualmente, la AFP se mide mediante ensayos inmunométricos de dos sitios (tipo sandwich) usando anticuerpos monoclonales y/o policlonales, con resultados similares a los RIA que los precedían. La mayoría de los ensayos comerciales están calibrados conforme a la Norma Internacional de la OMS (IS) 72/225. Los resultados clínicos se registran en unidades de masa (μg/L) o en unidades de kilo por litro de IS 72/225, para la cual 1 UI de AFP corresponde a 1,21 ng. El límite de referencia superior usado por la mayoría de los centros de tratamiento es 10-15 μg/L (8,3-12,4 kU/L). Según se informa, las concentraciones de AFP aumentan con la edad. El límite de referencia superior se incrementa de 11,3 μg/L en personas < 40 años a 15,2 μg/L en aquellas > 40 años (61). De manera ideal, se deberían establecer los valores de referencia por cada ensayo porque hay algunas variaciones en los resultados entre los métodos.
Microheterogeneidad de los hidratos de carbono de la AFP
La AFP es una glicoproteína y contiene 4% de hidratos de carbono de cadena simple que está N-ligada a la asparagina-232 del esqueleto proteico (62) (63). La microheterogeneidad de esta cadena de hidratos de carbono ha sido ampliamente investigada mediante el uso de la electroforesis de afinidad a la lectina (64-68) y el enfoque isoeléctrico (69-73). Se hallaron glucoformas distintivas características de un tejido maligno o benigno, lo que sugiere la posibilidad de mejorar la especificidad de la AFP para el CHC mediante la medición de una glucoforma específica para el CHC.
Las glucoformas de la AFP se pueden diferenciar según su afinidad de unión a la lectina (74-76). La AFP del suero del paciente con CHC, por ejemplo, se une más fuertemente a la concanavalina A que la AFP de los tumores de células germinales no seminomatosos, y ambos se unen más fuertemente a la lectina Lens culinaris (LCA) que la AFP de los pacientes con enfermedad hepática benigna. La afinidad a la LCA es ligeramente más alta para la AFP del CHC (AFP-L3) que la de los tumores de células germinales no seminomatosos (AFP-L2). Los campos de ensayos que están disponibles comercialmente miden específicamente las glucoformas AFP-L3 y la AFP-P4 (74)(76).
Numerosos estudios de Japón y de otros países asiáticos han demostrado que un incremento en la fracción de la AFP-L3 de la AFP sérica tiene una correlación más fuerte que la AFP sérica convencional con características histológicas adversas de CHC (por ej. mayor invasión de vena porta, tumor más avanzado, independientemente del tamaño) y predice resultados desfavorables (77-81). En un estudio que compara la medición de la AFP-L3 y la AFP en una población de estudio de los Estados Unidos (166 pacientes con CHC, 77 con enfermedad hepática crónica y 29 con masa hepática benigna), se descubrió que las concentraciones de AFP-L3 eran relevantes sólo en concentraciones de AFP entre 10 y 200 μg/L (82). Dentro de este rango, la AFP-L3 mostró una sensibilidad de 71% y una especificidad de 63% con un valor de corte de 10%. En un valor de corte > 35% la sensibilidad disminuyó a 33% pero la especificidad aumentó a 100%, permitiendo un diagnóstico confiable de un 10% adicional de casos de CHC que no hubieran sido diagnosticados usando sólo la AFP con un valor de corte de 200 μg/L.
En un estudio longitudinal prospectivo y multicéntrico de 2 años de duración en América del Norte, se comparó la AFP sérica con la AFP-L3 y la des-γ-carboxiprotrombina (DCP, un marcador tumoral para investigaciones del CHC) en 372 pacientes con hepatitis C (83), incluyendo 40 casos de CHC inicial, 34 casos de seguimiento de CHC y 298 casos inicialmente libres de CHC (83). La sensibilidad, especificidad y valores predictivos positivos/negativos fueron, respectivamente, 61%, 71%, 34% y 88% para la AFP (valor de corte de 20 μg/L) y 22%, 99%, 80% y 84% (valor de corte de 200 μg/L) comparados con 37%, 92%, 52% y 85% para la AFP-L3 sola (valor de corte de 10%) y 39%, 90%, 48% y 86% para la DCP sola (valor de cortede 7,5 μg/L; 83). Cuando se combinaron los tres marcadores, estas cifras aumentaron a 77%, 59%, 32% y 91% respectivamente. En pacientes con AFP elevada (20-200 μg/L), se halló alta especificidad para la AFP-L3 y la DCP (86,6% y 90,2%, respectivamente). De 29 pacientes con CHC y valores de AFP < 20 μg/L, 13 tenían concentraciones elevadas de AFP-L3 o DCP. Comparadas con la AFP total, las concentraciones normales de AFP-L3 y DCP tuvieron mayor correlación con la ausencia de CHC, mayor especificidad y valor predictivo negativo (83).
En un estudio prospectivo que compara la AFP-L3 y DCP con la AFP en 99 pacientes estadounidenses con CHC histológicamente confirmado, las tasas de sensibilidad fueron de 62%, 73% y 68% respectivamente, con la mayor sensibilidad (86%) obtenida cuando se combinaron los tres marcadores (84). La AFP-L3 estuvo relacionada de manera significativa con la invasión de la vena porta y los resultados en el paciente, sugiriendo que podría ser un marcador de pronóstico útil del CHC (84). Se investigó el uso de los mismos tres marcadores para predecir la recurrencia del CHC después de la ablación percutánea curativa en 416 pacientes con CHC, 277 de los cuales tuvo recurrencia durante el período de seguimiento (85). La AFP> 100 μg/L y AFP-L3> 15% pre- y post-ablación fueron indicadores significativos de recurrencia; por lo tanto, podrían complementar las modalidades de diagnóstico por imágenes en la evaluación de la eficacia del tratamiento (85). Recientemente se llevó a cabo un estudio importante y bien diseñado de caso control en siete centros médicos académicos de los Estados Unidos que comparaba la AFP, AFP-L3 y DCP (86). La cohorte de estudio incluyó a 417 pacientes con cirrosis y 419 con CHC-77 con BCLC muy temprana (BCLC 0) y 131 con enfermedad en estadio temprano (BCLCA). El análisis de la curva de diagnóstico (ROC) reveló que la AFP tuvo una sensibilidad mayor (67%) que la DCP o AFP-L3 en pacientes con enfermedad en estadio BCLC 0 (86). Se requiere investigación adicional para evaluar el valor de la AFP y los marcadores relacionados como criterios indirectos de valoración para obtener verdaderos resultados en materia de salud en los ensayos clínicos (87) (88).
LA AFP EN SCREENING Y DETECCIÓN TEMPRANA
Los pacientes cirróticos con concentraciones de AFP que sean persistentemente elevadas están en mayor riesgo de desarrollar CHC en comparación con aquellos con concentraciones de AFP que fluctúen o permanezcan dentro de los intervalos de referencia-29% vs. 13% vs. 2,4%, respectivamente; (6). Frecuentemente cuando el CHC se detecta durante un screening (89) se encuentran concentraciones de AFP séricas más bajas y los pequeños tumores CHC son AFP negativos en hasta 40% de los casos (90). La inmunotinción de AFP en CHC pequeños y bien diferenciados generalmente es negativa (91), haciendo que la AFP tisular resulte poco informativa. En estas instancias, los tumores sólo pueden ser detectables mediante ultrasonido (92). Las lesiones malignas no detectables por diagnóstico por imágenes son propensas a alcanzar un diámetro de 2 cm en aproximadamente 4-12 meses (93)(94). Para detectar tumores ≤ 2 cm de diámetro, se sugiere un intervalo de control de pacientes cirróticos de 6 meses, con el uso de AFP sérica y ultrasonido (95). Generalmente es difícil comparar los estudios debido a diferencias en el diseño de los mismos. Además, hay opiniones diversas sobre cómo la medición eficaz de la AFP contribuye con los programas de detección temprana o vigilancia (96). Se necesitan marcadores confiables para complementar el ultrasonido, porque la interpretación de éste último depende del operador y puede ser difícil de realizar en pacientes obesos o que tienen cirrosis subyacente (97).
En una revisión sistemática de las características de las pruebas de AFP para el diagnóstico del CHC en pacientes con VHC (98), sólo cinco de 1.239 estudios cumplieron todos los criterios de inclusión del autor (99-103). En estos cinco estudios, con el uso de un valor de corte de AFP de 20 μg/L, la sensibilidad varió de 41% a 65%, la especificidad de 80% a 94%, la relación de probabilidad positiva de 3,1 a 6,8 y la relación de probabilidad negativa de 0,4 a 0,6, demostrando en forma adicional que la AFP como prueba de screening tiene un valor limitado. En 19 de 24 estudios de pacientes con hepatitis C publicados de 1985 a 2002, la sensibilidad y especificidad de la AFP para el CHC fue de 45%-100% y 70%-95%, respectivamente, en puntos de corte entre 10 y 19 μg/L (104). Se informó que el ultrasonido tiene mayor sensibilidad (71%) y especificidad (93%) que la AFP sérica, pero el valor predictivo positivo del ultrasonido es bajo, cerca de 14% (30). Debido a que el éxito de la detección por ultrasonido es altamente dependiente de la capacidad del ultrasonógrafo, la investigación de pacientes con aumentos en la AFP sérica o nódulos sospechosos detectados en controles se realiza mejor en centros de derivación especializados.
La incidencia del CHC en pacientes con hepatitis crónica es más baja que en los pacientes con cirrosis, lo que puede disminuir el beneficio del screening en los primeros. Estudios japoneses sugieren que las diferencias en la historia natural de la hepatitis B y C significan que los pacientes con hepatitis B son más propensos a desarrollar CHC, inclusive cuando son jóvenes y se presentan asintomáticos (105).
En un estudio, fue necesario controlar a 1.069 pacientes infectados con el virus de la hepatitis B y con cirrosis demostrada para detectar 14 casos de CHC, de los cuales sólo seis estaban en un estadio suficientemente temprano como para poder ser tratados con cura quirúrgica (106). La frecuencia de detección de cáncer curable fue incluso menor en un estudio de 118 pacientes franceses con cirrosis A o B de la escala de Child-Pugh controlados en intervalos de 6 meses con ultrasonido, AFP y DCP. Sólo uno de los 14 casos detectados de CHC (7%) fue quirúrgicamente extirpable en el momento del diagnóstico (107). Sin embargo, otros estudios han demostrado beneficios en el screening de portadores de hepatitis B crónica para el CHC. Un estudio de screening prospectivo de una población de Alaska de 2.230 portadores con cirrosis que dieron positivo al antígeno de superficie de la hepatitis B (108) (109) demostró que 64%-87% de los CHC detectados estaban limitados a focos únicos y que 43%-75% de los tumores eran < 3 cm, lo que permitió una cirugía curativa en 29%-66% de los cánceres detectados (12) (110) (111). En otro estudio, se redujo significativamente el tamaño del tumor y la supervivencia mejoró (35% vs. 10% a los 30 meses) cuando se detectó el CHC mediante screening (112).
Hay pruebas de que el screening para la detección del CHC en poblaciones de alto riesgo puede ser rentable en regiones de alta prevalencia como Hong Kong (113) y que ofrece una ventaja de supervivencia, como se demostró en una población asiático-hawaiana asintomática con hepatitis B o C crónica y cirrosis (114) y también en un estudio italiano de pacientes cirróticos con CHC detectado mediante screening (115). Estas conclusiones están avaladas por resultados de un estudio de screening controlado y aleatorio de 18.816 pacientes de 35-59 años con infección por hepatitis B o antecedentes de hepatitis crónica (116) reclutados en la Shangai urbana entre 1993 y 1995. El screening semestral con AFP y ultrasonido redujo la mortalidad por CHC en 37%. Aunque los resultados de un estudio de screening de 5.581 portadores de hepatitis B entre 1989 y 1995 en el condado de Qidong demostró que el screening con AFP dio por resultado un diagnóstico más temprano del cáncer de hígado, aunque la ganancia del adelanto en el diagnóstico no resultó en una reducción total de la mortalidad (117). Parece probable que este hallazgo refleje diferencias en la terapia de los dos estudios, siendo que el 75% de los pacientes con CHC subclínico identificado en el estudio de Shangai recibió tratamiento radical mientras que sólo el 25% lo hizo en el estudio de Qidong (116).
Un estudio nacional de práctica en los Estados Unidos (118) documentó que la mayoría de las instituciones controlan de rutina el CHC en los pacientes con cirrosis, especialmente en aquellos con etiologías de alto riesgo. Muchos consideran que el screening sistemático de hígado con AFP y ultrasonido dos veces por año ofrece la mejor esperanza para el diagnóstico temprano del CHC en portadores sanos con antígeno de superficie de la hepatitis B positivo y factores de riesgo adicionales (por ej. hepatitis crónica activa, cirrosis) y en pacientes con cirrosis de cualquier etiología (119). El análisis de Markov ha demostrado claramente que en pacientes estadounidenses con cirrosis causada por la hepatitis C crónica, el screening de CHC es tan rentable como otros protocolos de screening aceptados (120). La AFP semestral y el ultrasonido anual proporcionaron la mayor ganancia en términos de años de vida ajustados por la calidad, mientras que mantuvieron una tasa de costo-efectividad < USD 50.000 por año de vida ajustado por la calidad. Los autores sugirieron que la AFP semestral con screening de TC anual podría ser incluso más costo-efectiva (120). Los resultados de una revisión sistemática y el análisis económico posterior indicaron que la AFP medida semestralmente y el ultrasonido realizado cada 6 meses constituyen la estrategia de control más efectiva en los pacientes de alto riesgo (121). Debido a los altos costos, sin embargo, los autores cuestionaron si se debería ofrecer de rutina el ultrasonido a aquellos con AFP sérica < 20 μg/L, en vista de la relación costo-beneficio, que depende de la etiología de la cirrosis.
Estas conclusiones en general son avaladas por los resultados de un reciente estudio modelo en el que se evaluaron la efectividad y la costo-efectividad de la vigilancia del CHC en cohortes separadas y combinadas de individuos con cirrosis debido a enfermedad hepática alcohólica, hepatitis B o hepatitis C (122). Se compararon los algoritmos que incluyen el uso de la AFP y/o del ultrasonido en intervalos de 6 y 12 meses. En la cohorte combinada, el modelo halló que la AFP y el ultrasonido realizados cada 6 meses eran más efectivos, triplicando el número de pacientes con tumores operables en el momento del diagnóstico y casi disminuyendo a la mitad la cantidad de muertes por CHC, en comparación con la ausencia de vigilancia. Según este informe, la estrategia más costo-efectiva incluiría un triage con mediciones de AFP cada 6 meses. Se concluyó que en el UK National Health Service, la vigilancia de individuos con cirrosis en alto riesgo de CHC debería ser considerada tanto eficaz como costo-efectiva (122).
Dado el amplio uso de las mediciones de AFP y ultrasonido hepático para realizar un screening prospectivo para la aparición del CHC en pacientes cirróticos, particularmente aquellos que son candidatos aptos para terapia curativa (109) (123) (124), hay una necesidad urgente de establecer y validar protocolos de seguimiento óptimos cuando se detectan nódulos sospechosos (10) (125) (126).
Las pautas clínicas basadas en la evidencia recientemente publicadas por un estudio japonés para el diagnóstico y tratamiento del CHC, diferencian el riesgo de CHC en pacientes con cirrosis como super-alto (cirrosis relacionada con hepatitis B/C) o alto (hepatitis crónica B/C o cirrosis hepática con causa diferente a la hepatitis B/C) (127)(128). Para el grupo de riesgo super-alto, se recomienda realizar el examen de ultrasonido y las mediciones de AFP, DCP y AFP-L3 con intervalos de 3-4 meses con TC o IRM dinámica cada 6-12 meses. Para el grupo de riesgo alto, se recomienda realizar un ultrasonido y mediciones de los marcadores tumorales cada 6 meses. Agregarle DCP o AFP-L3 se considera necesario dado que son marcadores de diagnóstico, mientras que la AFP es un marcador de riesgo (129) (130). La detección de una lesión nodular mediante ultrasonido y/o el aumento continuo de la AFP (> 200 μg/L), DCP [en unidades arbitrarias (UA) con 1 UA = 1 μg protrombina] (> 40 mUA/mL), o AFP-L3 (> 15%) requiere mayor evaluación mediante TC o IRM dinámica (127) (128).
La European Association for the Study of the Liver (EASL) recomendó que los nódulos de diámetros < 1 cm se controlen con una repetición de ultrasonido y AFP en 6 meses, que se agregue la biopsia e histología con aguja fina para investigar los nódulos de 1-2 cm (tasa falso-positivo 30%-40%) y que se empleen criterios adicionales de diagnóstico no invasivos (por ej., dos técnicas de imagen) para tumores > 2 cm (131). Las recomendaciones francesas publicadas en 2001 (132) indican que el diagnóstico del CHC debería basarse en el examen histopatológico de 1 o más muestras hepáticas obtenidas mediante cirugía abierta, laparoscopía o biopsia guiada por ultrasonido/TC (estándar) con la opción de aspiración con aguja fina para citología si la biopsia del hígado es imposible.
En un estudio retrospectivo estadounidense reciente en el cual los pacientes con lesiones hepáticas sospechosas de CHC fueron sometidos a aspiración con aguja fina y biopsia de núcleo, se compararon los resultados con aquellos de los métodos no invasivos comúnmente utilizados (133). Los pacientes con biopsias positivas tuvieron concentraciones de AFP sérica significativamente más altas que aquellos con resultados de biopsia negativos, aunque los dos grupos fueron similares en otros aspectos. Los resultados de la biopsia tuvieron mayor sensibilidad, especificidad y valor predictivo en comparación con los criterios de diagnóstico no invasivos. Los autores recomendaron un rol más importante de la biopsia guiada por imágenes de lesiones sospechosas >1 cm de tamaño para permitir la planificación adecuada del tratamiento, y observaron que los riesgos de la biopsia parecen pequeños y los beneficios potenciales, significativos (133).
Por supuesto que es esencial ser concientes de las advertencias del uso de la AFP, incluyendo las enfermedades benignas y malignas que pueden causar AFP sérica elevada y el hecho de que el valor dentro de los intervalos de referencia nunca excluya necesariamente al cáncer (99)(134). La AFP elevada detectada por una medición única puede ser pasajera (por Ej. que surja de un brote inflamatorio subyacente de la hepatitis viral crónica), mientras que las concentraciones elevadas pero estables disminuyen la probabilidad de que el CHC sea el agente causal. Las mediciones secuenciales de AFP sérica pueden brindar información útil, pero este hecho todavía está sometido a investigación y todavía no se ha validado completamente para la práctica clínica de rutina. Un patrón de crecimiento constante de AFP elevada debería ser siempre investigado rigurosamente usando ultrasonido y otras técnicas de imágenes. Si éstas arrojaran un resultado negativo inicialmente deberían repetirse para identificar cualquier posible cáncer hepático oculto (131).
En 2003, la British Society of Gastroenterology presentó guías para el uso de mediciones de marcadores tumorales seriales para realizar screening para la detección del CHC (26). El grupo de expertos concluyó que en los grupos de alto riesgo, el screening mediante ultrasonido abdominal y AFP comparado con ninguna vigilancia detectó CHC de menor tamaño. Dicha detección permite una mayor proporción de terapias curativas, siendo que la detección más temprana conduce a una mejor supervivencia a largo plazo y/o ahorro de costos. Se sugirió que el control de CHC debería estar restringido a hombres y mujeres con cirrosis por virus de hepatitis B o C o hemocromatosis genética y a hombres con cirrosis por cirrosis biliar primaria y cirrosis alcohólica (si es abstinente o es probable que cumpla con el tratamiento). Se consideró baja la probabilidad de que el CHC surja de una cirrosis de otra etiología. Se recomendó realizar vigilancia utilizando AFP y ultrasonido abdominal en intervalos de 6 meses, con equipos adecuados y operadores capacitados, elementos esenciales para el componente del ultrasonido. Se debe aconsejar a los pacientes sobre las implicaciones del diagnóstico temprano y su falta de beneficios demostrados (26).
Estas recomendaciones están en línea con las guías de la National Comprehensive Cancer Network (NCCN), que recomiendan la vigilancia usando AFP y ultrasonido en pacientes en riesgo de CHC (135). Entre los que se consideran en riesgo se encuentran los pacientes con cirrosis asociada con la hepatitis B o el alcohol con hemocromatosis genética, hepatitis autoinmune, esteatohepatitis no alcohólica, cirrosis biliar primaria o deficiencia de α1- antitripsina. Además, se recomienda el control de los individuos sin cirrosis portadores de hepatitis B o que tengan otros factores de riesgo (por ej., replicación viral activa, concentraciones altas de ADN del virus de la hepatitis B, antecedentes familiares de CHC, hombres asiáticos > 40 años, mujeres > 50 años, africanos < 20 años). La NCCN recomienda el diagnóstico por imágenes adicionales si la AFP sérica aumenta o luego de la identificación de un nódulo hepático durante el ultrasonido (135). La declaración de consenso de 2009 de la Asian Oncology Summit también recomienda el ultrasonido de hígado y la medición de las concentraciones de AFP cada 3-6 meses en todos los pacientes con cirrosis hepática, independientemente de la etiología, con la advertencia de que dicho control se establece mejor en cirrosis hepática relacionada con el virus de la hepatitis B, para el cual el LOE es relativamente alto (136). La AASLD actualmente recomienda el uso de la AFP para vigilancia pero sólo cuando el ultrasonido no se encuentra disponible (40). Esta organización también indica que el screening de detección de CHC debería ser "ofrecido en el contexto de un programa o proceso en el cual las pruebas de screening y los procedimientos de recuperación hayan sido estandarizados y en el cual los procedimientos de control de calidad estén en su lugar" (40).
De acuerdo con estas y otras recomendaciones (26)(131)(132)(135)(137) (Tabla II), la NACB apoya el uso de las determinaciones de AFP cada 6 meses y del ultrasonido abdominal para controlar en forma prospectiva el inicio del CHC en pacientes en alto riesgo, especialmente aquellos con cirrosis hepática relacionada con el virus de la hepatitis B o C.

AFP EN EL DIAGNÓSTICO
Las concentraciones de AFP séricas elevadas no son específicas del CHC porque también ocurren en el embarazo normal, en ciertas enfermedades hepáticas benignas y en algunos cánceres. Los procesos malignos sin CHC que puedan ocasionar altas concentraciones de AFP incluyen los tumores de células germinales no seminomatosos, para los cuales la AFP es un importante marcador tumoral con uso clínico bien establecido (138). La AFP también puede estar elevada en el cáncer de estómago, el cáncer del tracto biliar y el cáncer pancreático (139). Sin embargo, las concentraciones de AFP elevadas que exceden los 1.000 μg/L son raras en este tipo de malignidades, ocurriendo en < 1% de los casos.
Aproximadamente 20%-40% de los pacientes adultos con hepatitis o cirrosis hepática tienen concentraciones de AFP elevadas (>10 μg/L) (140). En estos pacientes, inicialmente se aceptaba una concentración de AFP entre 400 y 500 μg/L como el punto de decisión óptimo para diferenciar el CHC de la enfermedad hepática crónica (26)(136)(141-143). Sin embargo, un estudio japonés aconsejó un valor de corte óptimo de 150 μg/L según un análisis ROC (sensibilidad 54%, especificidad 95,9%, comparando los resultados para pacientes con CHC y enfermedad hepática crónica) (144). Utilizando la misma técnica ROC, un grupo italiano demostró la misma especificidad de 99,4% con valores de corte de 200 y 400 μg/L, pero con una sensibilidad más alta en el valor de corte inferior (99). Las pautas de la EASL 2001 indican que una AFP > 400 μg/L junto con la detección de un nódulo hepático sospechoso en diagnóstico por imágenes es diagnóstico de CHC (131). Esta guía está en línea con las recomendaciones del panel de la Asian Oncology Summit, que concluyó que las imágenes características de TC o IRM dinámica, independientemente del tamaño del tumor, son suficientes para el diagnóstico de CHC y excluyen la necesidad de realizar una biopsia, con AFP > 400 μg/L diagnóstica en pacientes con cirrosis hepática o hepatitis crónica (136). Este grupo también recomendó evitar la biopsia de aguja cuando la cirugía curativa sea posible. Los paneles de expertos japoneses (131) y de la AASLD (40) indican que en pacientes con nódulos hepáticos sospechosos en diagnóstico por imágenes, las concentraciones de AFP > 200 μg/L también son sospechosas y deberían ser investigadas. Después de la exclusión de la inflamación hepática, un aumento sostenido de la AFP es indicador de CHC y debería dar lugar a mayores estudios por imágenes del hígado, mientras que los resultados estables o en disminución hacen que sea menos probable.
Las concentraciones de AFP en circulación en pacientes con CHC fluctúan desde dentro de los intervalos de referencia hasta tan alto como 10 × 106 μg/L (es decir, 10 g/L), con concentraciones previas al tratamiento > 1.000 μg/L en aproximadamente 40% de los pacientes (145). Se informó que la AFP es más alta en pacientes con CHC que surge a partir de condiciones virales crónicas comparadas con aquellas de la enfermedad hepática alcohólica (146) y en pacientes más jóvenes (147) y masculinos (147). En un estudio de cohorte de 239 pacientes con hepatitis crónica, 277 con cirrosis y 95 con CHC, la AFP arrojó sensibilidades de CHC de 79% y 52,6% en puntos de decisión de 20 μg/L y 200 μg/L, respectivamente, con especificidades correspondientes de 78% y 99,6% (148). De acuerdo con algunos investigadores japoneses (149), cualquier valor de AFP circulante > 10 μg/L en pacientes con enfermedad hepática crónica debería ser considerado sospechoso de CHC y dar lugar a mayores investigaciones (por ej., usando pruebas de lectina AFP-L3 [LCA] o AFP-P4 [EPHA] y diagnóstico por imágenes). Estos investigadores sugieren un punto de decisión más bajo de 10 μg/L en lugar de 20 μg/L para tomar en cuenta las mejorías en el diagnóstico por imágenes que han conducido a una mayor detección de CHC cuando la AFP es < 20 μg/L. En Japón, por ejemplo, el porcentaje de pacientes con CHC con concentraciones de AFP < 20 μg/L en la consulta aumentó de 3,6% en 1978 a 38,1% en 2000. Desde 2001 a 2003, después de un cambio en el valor de corte de AFP de < 15 μg/L, 36,4% de los pacientes con CHC habían aumentado las concentraciones de AFP (127). Un informe anterior acerca de que los individuos japoneses saludables no tienen concentraciones de AFP > 10 μg/L (150) apoyó la introducción de un corte más bajo, pero este hallazgo puede aplicarse solamente a la población estudiada.
Las pautas japonesas indican que el CHC se puede diagnosticar por imágenes (ultrasonido mejorado por contraste de TC/IRM dinámica) u otras técnicas (hipervascularidad en la fase arterial y lavado en la fase venosa portal (127) (128)). Los aumentos continuos de AFP (> 200 μg/L) y/o DCP (> 40 mUA/mL) y/o AFP-L3 (> 15%) son altamente sugestivos de CHC típico, incluso en ausencia de evidencia de un nódulo hepático aparente por ultrasonido (127), y debería indicar el uso de TC o IRM dinámica (128).
De acuerdo a pautas recientes de la AASLD, la vigilancia/screening en pacientes de riesgo de CHC debería realizarse usando ultrasonidos en intervalos de 6-12 meses y no usar la AFP sola a menos que el ultrasonido no esté disponible (40), mientras que las guías de la NCCN recomiendan un screening periódico con ultrasonido y AFP cada 6-12 meses (135). Durante la detección de un nódulo < 1 cm por ultrasonido, el panel de la AASLD recomienda un seguimiento por ultrasonido en intervalos de 3-6 meses, revirtiendo el control de rutina si no hay crecimiento después de un período de hasta 2 años (40). En contraste, las guías de la NCCN recomiendan el control mediante imágenes por TC/IRM/ultrasonido cada 3-4 meses para nódulos < 1 cm, revirtiendo el control de rutina si el nódulo no aumenta su tamaño durante 18 meses (135). Los nódulos de 1-2 cm que se detectan en hígados cirróticos por ultrasonido deben ser investigados mediante dos estudios dinámicos (por ej. TC, IRM) y tratados como CHC si su apariencia es consistente con este diagnóstico, pero si no es característica, la lesión debería ser sometida a biopsia.
Para un nódulo > 2 cm en el diagnóstico inicial con características de CHC típicas (por ej., aumento arterial clásico en TC o IRM trifásica) o en casos en los cuales la AFP es > 200 μg/L, los resultados se pueden considerar como diagnóstico de CHC y la biopsia ser innecesaria; pero si la lesión no es característica, o el hígado no es cirrótico, se recomienda realizar la biopsia. Para lesiones pequeñas que son negativas en una biopsia, se recomienda el seguimiento de ultrasonido o TC en intervalos de 3 a 6 meses, con repetición de la biopsia si la lesión se agranda pero permanece atípica. Las lesiones que ocupan espacio con hipoperfusión por la sangre portal se consideran un signo temprano de CHC, inclusive en ausencia de un aumento coincidente en la AFP circulante.
La EASL (131), la British Society of Gastroenterology (26), el European Group on Tumor Markers (137) y la NCCN recomiendan el uso de AFP como anexo para el diagnóstico de CHC. Estas recomendaciones son avaladas por el Panel de la NACB, que también recalca la importancia de las mediciones de AFP sérica junto con la consideración de los aumentos continuos en la AFP incluso en bajas concentraciones (Tabla II).

AFP EN EL PRONÓSTICO
El sistema TNM (151) y la clasificación de Okuda (152) son los sistemas de estadificación más frecuentemente utilizados para el CHC. Las clasificaciones pronósticas de Japón (153), Francia (154), Italia (32)(155), España (156) (157) y China (158) también han sido publicadas [ver también (159) (160). De estos, el sistema de estadificación BCLC español mostró la mejor estratificación de pronóstico (161) y también fue adoptado en las pautas de la AASLD (40). La mayoría de estos sistemas incluyen como factores principales de pronóstico la gravedad de la enfermedad hepática subyacente, el tamaño del tumor, la extensión del tumor a las estructuras adyacentes y la presencia de metástasis <zref>152,<ths>155<zrefx>. De acuerdo con las pautas de la AASLD (40), para la evaluación óptima del pronóstico de pacientes con CHC, el sistema de estadificación debería incluir el estadio del tumor, la función hepática y el estado físico y considerar la expectativa de vida, factores incluidos en el sistema BCLC español.
El sistema de estadificación chino (corte de AFP de 500 μg/L) (158) y dos sistemas de estadificación europeos incluyen a la AFP. El sistema francés incluye el índice de Karnofsky, obstrucción de la vena portal ultrasonográfica y bilirrubina sérica, fosfatasa alcalina y AFP (corte de 35 μg/L) (154). En base a la puntuación, los pacientes se clasifican como en riesgo de muerte bajo, moderado o alto, con tasas de supervivencia a 1 año de 72%, 34% y 7%, respectivamente. Otra clasificación, propuesta por el programa italiano Cancer of the Liver (155), incluye el estadio Child-Pugh, morfología, trombosis de la vena porta y AFP sérica (corte de 400 μg/L). Mediante el uso de un sistema de puntuación simple, los pacientes son asignados a una de siete categorías con tasas de supervivencia promedio validadas (155). Ambas clasificaciones incorporan a la AFP como indicador de diseminación y carga del tumor, la diferenciación celular y el potencial agresivo. Con el objetivo de mejorar los sistemas disponibles para la clasificación del riesgo postoperatorio, recientemente se ha desarrollado un nomograma basado en variables clínico-patológicas incluyendo la AFP sérica, la edad del paciente, el tamaño del tumor y el estado de los bordes, pérdida de sangre postoperatoria, presencia de lesiones satélites e invasión vascular. Según se informa, el nomograma permite la predicción precisa de supervivencia postoperatoria y estratificación de riesgo en pacientes que son sometidos a la resección hepática para HCC y que actualmente se encuentran en evaluación (162).
Se ha sugerido que considerar la AFP y la fosfatasa alcalina, la puntuación Child-Pugh y la ausencia o presencia de ascitis podría mejorar la predicción de resultados (46) (154) (155). Un estudio italiano de factores de pronóstico en 176 pacientes con CHC demostró que albúmina baja (< 33 μg/L), bilirrubina alta (> 22,5 μmol/L), AFP elevada (> 32,5 kU/L), trombosis de la vena porta y una lesión intratable fueron factores de riesgo independientes para una sobrevida desfavorable (163). La sobrevida dependió más fuertemente del grado de disfunción hepática, presencia de infección por virus de la hepatitis B, tipo de diagnóstico y agresividad del tumor. Un estudio japonés reciente de factores de pronóstico que influyen en la sobrevida después de la resección del hígado en pacientes con CHC demostró una mejoría en los resultados y tasas de mortalidad operatoria durante la última década (164). La edad, el grado de daño hepático, la concentración de AFP, la dimensión máxima del tumor, la cantidad de tumores, la extensión intrahepática del mismo, la metástasis extrahepática, la invasión de la vena porta y hepática, la curabilidad quirúrgica y los bordes quirúrgicos libres fueron factores de pronóstico en pacientes con CHC sometidos a resección hepática (164).
Los grandes estudios que utilizan análisis multivariados confirman que las concentraciones elevadas de AFP predicen un pronóstico desfavorable en comparación con los casos negativos para AFP en CHC (32)(154)(165). En un estudio retrospectivo de 309 pacientes con CHC estratificado según concentraciones de AFP previas al tratamiento (< 20, 20-399, o ≥ 400 μg/L), los pacientes con concentraciones de AFP más altas tendieron a tener tumores más grandes, pero no hubo correlación con el estadio Okuda, el grado de diferenciación del tumor o la metástasis extrahepática (166). En contraste, un estudio multicéntrico italiano grande más reciente, que utilizó los mismos tres grupos de AFP en 1.158 pacientes con CHC (167) reveló una baja sensibilidad (54%) de AFP en el diagnóstico de CHC, pero confirmó su valor pronóstico demostrando que tenía una correlación significativa con el tamaño del tumor, la focalidad de la lesión, el estadio TNM y Okuda, la puntuación Edmonson y la supervivencia (P < 0,0001) en pacientes tratados y no tratados.
Según otros autores (168) (169), la AFP, así como también el tamaño del tumor, parece ser un indicador independiente de supervivencia. La supervivencia de pacientes con AFP sérica > 10.000 μg/L en el diagnóstico fue significativamente más corta que en aquellos con AFP < 200 μg/L (tiempo de supervivencia promedio de 7,6 vs. 33,9 meses, respectivamente)(170). Las concentraciones de AFP > 1.000 μg/L predicen un pronóstico relativamente más desfavorable, incluso después de intentar una resección curativa (70). Se requieren concentraciones de AFP sérica < 12.000 μg/L para cumplir con los criterios del Reino Unido para el transplante de hígado (171).
El tiempo de duplicación de la AFP también ha sido indicado como un factor de pronóstico importante (172). La persistencia de una fracción de AFP-L3 positiva después de la intervención también ha sido informada como indicadora de enfermedad residual o recurrente (77). La NACB apoya el uso de la concentración de AFP sérica previa al tratamiento para pronóstico, en combinación con otros factores de pronóstico (Tabla II).

AFP EN EL CONTROL DE PACIENTES DESPUÉS DEL TRATAMIENTO
Para pacientes con concentraciones de AFP elevadas antes de la terapia, el tratamiento de control de CHC mediante el uso de determinaciones de AFP seriadas es un procedimiento bien aceptado. Después de la extracción completa del tumor, las concentraciones de AFP generalmente disminuyen, con una vida media de 3,5-4 días. La extracción incompleta resulta en una media vida más alta, asociada con una supervivencia más desfavorable (166)(172), mientras que la incapacidad de que la AFP se normalice implica malignidad residual o daño hepático grave. La determinación de la fracción de AFP-L3 puede ayudar a diferenciar entre estas dos condiciones (81)(142)(173). Sin embargo, la normalización de la AFP no indica necesariamente la ausencia total de la enfermedad. Puede haber recurrencia después del transplante, incluso cuando la AFP es estable y se encuentra dentro de los límites normales (168)(172)(174), posiblemente reflejando la presencia de una micrometástasis demasiado pequeña como para producir concentraciones séricas mensurables.
Los cambios en las concentraciones de AFP también reflejan una respuesta del tumor después de la quimioterapia, con mayor sobrevida en pacientes que muestran una disminución significativamente prolongada de la AFP respecto de aquellos con concentraciones en lento aumento (175) (176). En pacientes que reciben nuevas terapias sistémicas combinadas efectivas (177), 75% ha demostrado disminuciones drásticas en la AFP sérica, con concentraciones en proceso de normalizarse completamente en algunos pacientes. Se descubrió una enfermedad progresiva en pacientes con aumento continuo de AFP y tiempos de duplicación entre 6,5 y 112 días (promedio 41 días), nuevamente en correlación con la sobrevida (172). Se observaron resultados similares después de la radioterapia para tumores hepáticos primarios y secundarios. Las disminuciones en marcadores tumorales reflejaron la regresión del tumor con mayor consistencia que los cambios posteriores en su tamaño y volumen según fuera determinado por la TC (178). Las discrepancias entre el marcador tumoral y los resultados de las imágenes pueden deberse a la fibrosis residual y otros factores que pueden complicar la interpretación de la TC (178).
Un estudio reciente aleatorio en fase III de quimioterapia sistémica en pacientes con CHC evaluó los resultados clínicos y radiológicos e incluyó mediciones de AFP seriadas, en forma prospectiva (179). En 117 pacientes con AFP sérica inicialmente elevada (valor de corte 20 μg/L) y una respuesta de AFP (≥ 20% disminución) después del segundo ciclo de quimioterapia, 47 habían mejorado su sobrevida y 70 no respondieron a la AFP (13,5 vs. 5,6 meses; P < 0,0001). Las concentraciones de AFP estuvieron fuertemente asociadas con la respuesta radiológica (P < 0,0001) y también con la sobrevida (análisis multivariado: razón de riesgo de 0,413, P < 0,0001). Por lo tanto, se concluyó que en pacientes con CHC sometidos a quimioterapia sistémica, las determinaciones de AFP en serie pueden ser útiles para el pronóstico y para el control de la respuesta al tratamiento, así como también para brindar un marcador indirecto para la evaluación de nuevos agentes terapéuticos (179). De manera similar, los autores de un estudio reciente del Massachusetts General Hospital Cancer Center y del Harvard Medical School concluyeron que el cambio en la AFP sérica durante el tratamiento puede servir como marcador indirecto para los resultados clínicos en pacientes con CHC avanzado sometidos a terapia sistémica (180).
Según la French Strength of Recommendation (SOR) (132), no hay consenso sobre los patrones o modalidades de seguimiento más que el examen clínico y los planes de vigilancia que pueden incorporar ultrasonido, mediciones de AFP, TC abdominales, radiografías de tórax y/o IRM, con la elección y sincronización óptimas de éstas dependiendo de las opciones de tratamiento. La NCCN es más específica y recomienda el seguimiento postratamiento de pacientes con CHC que incluye diagnóstico por imágenes cada 3-6 meses durante 2 años y luego anualmente, con mediciones de AFP (si inicialmente es elevada) cada 3 meses durante 2 años y luego cada 6 meses (135). De manera similar, ESMO recomienda que los pacientes sometidos a la resección curativa deberían ser controlados con diagnóstico por imágenes del hígado y mediciones de AFP durante 2 años con intervalos de 3 a 6 meses y luego anualmente, porque se puede ofrecer la terapia curativa a una minoría de pacientes después del relapso (4). Después del transplante de hígado, el seguimiento debe ser más frecuente (es decir, mensualmente cada 6 meses, luego una vez cada 3 meses hasta un año después del transplante, luego dos veces al año hasta los dos años posteriores y luego todos los años)(4).
De acuerdo con otros grupos de expertos (131) (132)(135), la NACB recomienda determinaciones en serie de la AFP sérica (si es elevada antes del tratamiento) para controlar la eficacia del tratamiento, el curso de la enfermedad y la recurrencia, y avala la frecuencia de las mediciones recomendadas por la NCCN (135).

MARCADORES TUMORALES DIFERENTES A LA AFP
Des-γ-Carboxiprotrombina (DCP)
La DCP, también conocida como protrombina producida por la ausencia de vitamina K o antagonismo II (PIVKA II), es una protrombina anormal carente de actividad coagulante y potencial marcadora del CHC.
Principalmente desarrollada e investigada en Japón, la DCP fue descripta por primera vez en los Estados Unidos en 1984 (181) y fue revisada críticamente allí en 1993 (182). Un equipo EIA comercialmente disponible desde Japón ha dominado el mercado de las pruebas de DCP. Se ha mejorado significativamente la sensibilidad de este método desde 1996, y actualmente es de 10 mkU/L.
Una cantidad de investigaciones publicadas han informado sensibilidades de la DCP de 54% a 70% para el diagnóstico del CHC en un punto de decisión de 40 mAkU/L, con especificidades correspondientes en pacientes cirróticos entre 87% y 95%. La AFP probada simultáneamente en los mismos pacientes ha demostrado, en un punto de decisión de 20 μg/L, 47%-72% de sensibilidad y 72%-86% de especificidad. La sensibilidad combinada de DCP/AFP fue de aproximadamente 80% (183-186). Las sensibilidades DCP, AFP y DCP/AFP combinadas para CHC solitarios (< 2 cm) fueron de 30%- 53%, 13% y 57% respectivamente, y para tumores más grandes (> 3 cm) fueron de 78%-81%, 49%-69% y 84%- 94% respectivamente (183)(184) (186). La sensibilidad de ambos marcadores fue mejor para tumores diferenciados en forma moderada o deficiente (DCP, 68%; AFP, 61%; DCP/AFP, 85%; n = 41) que para tumores bien diferenciados (DCP, 13%; AFP, 33%; DCP/AFP, 40%; n = 15) (186). Las concentraciones de DCP y AFP guardaron correlación con el tamaño y la estadificación del tumor, pero no significativamente entre sí.
Un estudio transversal de control de casos que comparó la AFP sérica y la DCP en una población estadounidense ha confirmado la aparente superioridad de la DCP como marcador tumoral del CHC (187). El estudio incluyó 48 adultos sanos, 51 pacientes con hepatitis crónica (mayormente hepatitis C), 53 individuos con cirrosis compensada y 55 personas con CHC demostrado. Con el uso del análisis ROC, se descubrió que la DCP funcionaba mejor que la AFP en la diferenciación del CHC de cirrosis (90% vs. 77% de sensibilidad, 91% vs. 71%, de especificidad, 85% vs. 81% de valor predictivo positivo, 90% vs. 74% de valor predictivo negativo, 0,921 vs. 0,815 de área debajo de la curva ROC). No hubo mejoras sobre la DCP sola al combinar a los 2 marcadores.
También se informó que la DCP tiene importancia en el pronóstico. En un estudio de pacientes con CHC tratados mediante inyección de etanol percutánea o terapia de coagulación por microondas, el análisis multivariado demostró que después de la clasificación histológica y la diferenciación del tumor, la DCP fue el factor predisponente más fuerte para el desarrollo posterior de invasión de la vena porta (188), mientras que los resultados del análisis ROC sugirieron que es un predictor efectivo de la recurrencia de CHC después de la resección (189). En otro estudio, se categorizaron 237 pacientes con CHC en cuatro grupos según las concentraciones de DCP (menor o mayor que 62,5 mAkU/L) y AFP (menor o mayor que 100 μg/L) (190). Los 22 pacientes con AFP baja y DCP alta fueron predominantemente hombres y tenían lesiones grandes pero pocos nódulos. El resultado fue particularmente deficiente en los pacientes que tenían concentraciones altas de DCP y AFP (190). De acuerdo con un informe más reciente que compara las determinaciones de AFP sérica y DCP en 1.377 pacientes con CHC y 355 pacientes con enfermedad hepática crónica, la utilidad de la DCP fue más baja en tumores más pequeños (< 3 cm de diámetro) que en los más grandes (> 5 cm de diámetro) (191).
Un análisis retrospectivo de 199 pacientes con CHC de estadio temprano en pacientes cirróticos Child-Pugh A tratados mediante resección o ablación por radiofrecuencia (RFA) demostró tasas de sobrevida similares de 3 y 5 años (90%/79% vs. 87%/75%) (192). Las tasas de sobrevida sin recurrencia de tumor en uno y tres años fueron más altas en los pacientes tratados mediante resección (83%/51% vs. 83%/42% para RFA; P = 0,011) (192). Con el análisis multivariado, se halló que el tiempo de protrombina ≥ 80% es un factor de pronóstico independiente para el grupo sometido a la resección mientras que el recuento de plaquetas ≥ 100.000 y la concentración de DCP < 100 UA/L fueron pronósticos para el grupo de RFA. En concentraciones de DCP≥ 100 UA/L, el procedimiento de tratamiento se convirtió en un factor de pronóstico significativo para la sobrevida. Estos resultados sugieren que la concentración alta de DCP refleja agresividad biológica y que la resección quirúrgica es más ventajosa en estos pacientes que el tratamiento de RFA. Se ha investigado el valor pronóstico de las concentraciones de AFP previas al tratamiento (valor de corte de 400 μg/L), AFP-L3 (de 15%) y DCP (de 100 AU/L) en pacientes con CHC después del tratamiento curativo mediante hepatectomía (n=345) y comparado con la ablación térmica locorregional (n=456; 173). Los resultados de los análisis multivariados en pacientes con hepatectomía indicaron que ningún marcador tumoral se encontraba asociado con una sobrevida disminuida. En pacientes que habían sido sometidos a ablación térmica locorregional, la elevación de la AFP-L3 (P= 0,0171) o DCP (P= 0,0004) estuvo significativamente asociada con la sobrevida disminuida y la DCP también estuvo asociada con una tasa de recurrencia elevada (P < 0,0001).
Una investigación de AFP, AFP-L3 y DCP en 240 pacientes con hepatitis B o C (144 con CHC, 47 con hepatitis crónica y 49 con cirrosis) en valores de corte óptimos según el análisis ROC (DCP, 84 AU/L; AFP, 25 μg/L; AFP-L3, 10%) arrojó tasas de sensibilidad, especificidad y valor predictivo positivo de 87%, 85% y 86,8% para la DCP; 69%, 87% y 69,8% para la AFP; y 56%, 90% y 56,1% para la AFP-L3 (193). Las concentraciones de DCP estaban debajo del valor de corte en todos los casos sin CHC, pero elevadas en todos los casos de CHC incluyendo aquellos con lesiones únicas. La DCP guardó correlación con el tamaño del tumor, las concentraciones altas de AFP con el tipo difuso de CHC y los tres marcadores con el CHC metastático. Los autores recomendaron el uso de rutina de la DCP para la detección del CHC.
Las concentraciones falso-positivas elevadas de DCP se encuentran en pacientes con ictericia obstructiva grave debido a colestasis intrahepática o en condiciones en que la acción de la vitamina K está disminuida (por ej., en personas con deficiencia continua de vitamina K y aquellos que han ingerido warfarina y antibióticos de amplio espectro) (194). A pesar de estas limitaciones, la DCP es un marcador emergente prometedor con un potencial considerable.
Glipican 3
El glipican 3 (GPC-3), inicialmente conocido como MXR7 (195), es otro promisorio marcador tisular y sérico nuevo para el CHC. El gen glipican 3 (GPC3) codifica un miembro de la familia glipican de proteoglicanos heparínsulfato de la superficie celular anclados por glicosilfosfatidilinositol (196). El GPC-3 fue detectado por primera vez a través de su ARNm, elevado en 75% en muestras de tejidos de pacientes con CHC primario y recurrente pero en sólo 3,2% de las muestras de tejido hepático normal (195). Estos datos luego fueron confirmados en forma inmunohistoquímica (196) (197). Se encontraron concentraciones de ARNm GPC- 3 elevadas en el suero de pacientes con CHC (195). La sensibilidad excedió la de la AFP (88% vs. 55%) para todo el grupo de pacientes con CHC analizado, así como también para aquellos con tumores de CHC < 3 cm (77% vs. 43%). En un estudio posterior de 34 pacientes con CHC (196), la sensibilidad fue un tanto más baja (53%) y similar a la de la AFP (54%). Sin embargo, la especificidad fue excelente, sin elevaciones significativas en donantes de muestras sanas o en pacientes con hepatitis aguda, y sólo en uno de los 20 pacientes con hepatitis crónica o cirrosis. La sensibilidad combinada de los dos marcadores fue de 82%. Ningún marcador tuvo correlación con el otro.
Aunque otro grupo demostró la presencia del extremo C terminal en suero (198), un informe reciente de la proteína GPC sugiere que el único fragmento presente en la circulación es el amino terminal, que constituye el marcador serológico soluble GPC-3 (sGPC- 3)(199). Con el uso de una prueba ELISA con anticuerpos monoclonales altamente específicos para analizar el suero de 69 pacientes con CHC, 38 pacientes con cirrosis hepática y 96 adultos sanos, el análisis ROC arrojó tasas de sensibilidad/especificidad de 51%/90% para sGPC-3 (punto de corte de 2 μg/L) comparable con los de la AFP (55%/90%; corte de 20 μg/L). La sensibilidad de los dos marcadores en un subconjunto de CHC de estadio temprano fue esencialmente la misma, y no hubo correlación entre sGPC- 3 y AFP en los 69 pacientes que tenían CHC. La sensibilidad del marcador combinado fue de 72%. Este estudio preliminar sugiere que la sGPC-3 puede ser prometedora y que amerita realizar estudios clínicos más grandes para investigar su potencial.
OTROS MARCADORES SÉRICOS PARA EL CÁNCER HEPÁTICO
Se han informado muchos otros marcadores séricos para el CHC (Tabla I). Algunos grupos han sugerido que la detección previa y posterior al tratamiento de las células de CHC circulantes mediante transcripción inversa (RT)-PCR de ARNm AFP es útil en la predicción de recurrencia de CHC y resultados desfavorables (200)(201), aunque otros investigadores han cuestionado su valor (202-204). Otras técnicas bajo investigación son el perfil genético, el estudio de los transcriptomas (205-207), el análisis de los proteomas (208) (209) y la determinación de ácidos nucleicos libres (210) y anormalidades epigenéticas (por ej. p16 hipermetilada) en el suero o el plasma (211). También se están explorando las implicaciones de pronóstico de la hipermetilación de CpG e hipometilación del ADN (212), perfil del microARN (213) y exploración de células madre de cáncer hepático (214). Se han identificado cincuenta genes marcadores sobrerregulados del CHC, que son candidatos potenciales a marcadores tumorales de CHC asociado al virus de la hepatitis C mediante el uso de un análisis de matrices de ADNc de muestras de hígado quirúrgico de pacientes infectados con el virus de la hepatitis C (215).
El panel de la NACB no recomienda el uso de ningún biomarcador relacionado con el CHC excepto la AFP para el control de rutina de pacientes que padecen, o están en riesgo de padecer, CHC. La NACB sí apoya futuras evaluaciones de la utilidad clínica de marcadores potenciales para los cuales hay evidencias publicadas en aumento (por ej. AFP-L3, DCP y GPC-3) en estudios clínicos aletorios prospectivos bien diseñados.

PUNTOS CLAVE: MARCADORES TUMORALES EN CHC
El CHC es uno de los cánceres más comunes en el mundo y frecuentemente es precedido por la hepatitis B o C crónica o la enfermedad hepática alcohólica. Si el tratamiento de estas enfermedades se instituye en forma temprana, el riesgo de CHC puede disminuir o ser eliminado. En pacientes que ya han desarrollado CHC, la resección quirúrgica o el transplante con intención curativa requiere detección local temprana de lesiones pequeñas. La utilidad clínica de la medición de AFP, junto con el ultrasonido y otras técnicas de imágenes más sensibles, ya está bien establecida para esta aplicación, mientras que otros marcadores tumorales requieren más investigación. Los desarrollos futuros en genética molecular y análisis proteómico pueden conducir a un diagnóstico más temprano y a un tratamiento más efectivo de los pacientes con CHC.
Marcadores tumorales en cáncer de vejiga
Antecedentes
Aproximadamente 71.000 casos nuevos de cáncer de vejiga se diagnostican por año en los Estados Unidos, y alrededor de 14.000 personas mueren de esta enfermedad (216). La prevalencia de cáncer de vejiga en los Estados Unidos se estima en casi 500.000 casos. En hombres se producen aproximadamente el doble de casos que en mujeres, siendo el tabaquismo la principal causa (217). Entre los factores de riesgo se encuentran la exposición a carcinógenos industriales e infecciones crónicas con Schistosomiasis haematobium.
El síntoma más común del cáncer de vejiga es hematuria intermitente (80%-85% de los pacientes). Otros síntomas del tracto urinario son mayor frecuencia de micción, urgencia y disuria (15%-20% de los pacientes). Generalmente se establece el diagnóstico por medio de evaluación cistoscópica, alertada por hematuria o síntomas en el tracto urinario, y por biopsia. En algunos casos, la citología urinaria es positiva para células tumorales. El cáncer de vejiga se estadifica de acuerdo al grado de invasión del tumor en la pared de la vejiga (218). El carcinoma in situ (estadio Tis) y los estadios Ta y T1 se agrupan como cánceres de vejiga invasivos no musculares porque están restringidos a la cubierta epitelial interior de la vejiga y no involucran a la pared muscular. De los tumores invasivos no musculares, los tumores de estadio Ta están confinados a la mucosa, mientras que los tumores de estadio T1 invaden la lámina propia. Se considera que los tumores T1 son más agresivos que los tumores Ta (219). Los tumores invasivos musculares (estadios T2, T3, y T4) se extienden hacia adentro del músculo (estadio T2), la capa adiposa perivesical posterior al músculo (estadio T3), y órganos adyacentes (T4). Los tumores metastásicos afectan nódulos linfáticos (N1-3) u órganos distantes (M1).
El tipo más común de célula de cáncer de vejiga es el carcinoma de célula transicional, a pesar de que se producen también adenocarcinomas, carcinomas de células escamosas y sarcomas. La morfología celular de los tumores de vejiga invasivos no musculares se clasifica de acuerdo con el grado de diferenciación celular. La clasificación consiste en tumores bien diferenciados (grado 1), moderadamente diferenciados (grado 2), y pobremente diferenciados (grado 3). La clasificación de la morfología celular es importante para establecer un pronóstico debido a que los tumores de grado 3 son los más agresivos y los que más probablemente resulten invasivos. Se está muy a favor del uso de la clasificación de la OMS de 2004 porque facilita el diagnóstico uniforme de los tumores (220). Un sistema de clasificación modificado (WHO International Society of Urological Pathology 1998), que está siendo utilizando con más frecuencia (221), elimina los grados numéricos y categoriza a la mayoría de los cánceres de vejiga como de bajo o alto grado.
La heterogeneidad de los tumores urológicos -en términos tanto de origen histológico como de comportamiento clínico- significa que los parámetros clínicos como el grado o estadio del tumor no son lo suficientemente precisos como para predecir el comportamiento biológico o para guiar el tratamiento de manera confiable, especialmente en los casos de alto riesgo (223-225). Se necesitan de manera urgente nuevos marcadores para ayudar en el diagnóstico, evaluar el pronóstico, identificar el tratamiento óptimo, y monitorear la progresión de los cánceres urológicos.
El cáncer de vejiga puede ser considerado una enfermedad genética causada por la acumulación progresiva de factores genéticos y epigenéticos (226-228). Los tumores de vejiga invasivos no musculares son tratados generalmente por resección transuretral de la vejiga con o sin tratamientos intravesicales con inmunoterapia con bacilo de Calmette-Guérin o quimioterapia intravesical. Los tumores invasivos musculares son tratados generalmente por cistectomía, o con terapias de preservación vesical que consisten en quimioterapia y radiación. Los pacientes que tienen enfermedad metastásica requieren quimioterapia sistémica con múltiples agentes anticancerígenos (229). Una clara comprensión de las vías de progresión del cáncer facilita el desarrollo de terapias con drogas contra objetivos tumorales específicos (225).
A la mayoría de los pacientes con cáncer de vejiga se les diagnostican tumores invasivos no-musculares. A pesar de que estos tumores pueden extirparse completamente, existe un alto riesgo de recurrencia; 50%-70% de estos pacientes van a tener recurrencia del tumor dentro de los 5 años siguientes. Con vigilancia médica intensiva, las tasas de supervivencia a 5 años para aquellos pacientes oscilan entre 95% y 75% para los tumores Ta y T1 respectivamente. Sin embargo, casi el 25% de los pacientes con tumores no-invasivos Ta y T1, con el tiempo desarrollarán enfermedad invasiva. La tasa de supervivencia a 5 años disminuye con la invasividad del tumor y la presencia de metástasis. Los pacientes con tumores en estadio T2 tienen una tasa de supervivencia a 5 años del 60%, pero solamente 35% de los pacientes con tumores T3 y 10% de los pacientes con tumores metastásicos T4 sobreviven 5 años (218).
Es por lo tanto necesario realizar vigilancia durante toda la vida de los pacientes con cáncer de vejiga a los que inicialmente se les diagnostica enfermedad invasiva no muscular. Los protocolos actuales de control de pacientes generalmente consisten en evaluaciones citoscópicas pautadas regularmente, generalmente junto con citología urinaria, realizada cada 3 meses durante los primeros 2 años de seguimiento, dos veces al año durante los años 3 y 4, y una vez al año de ahí en adelante, hasta que se documente la recurrencia de la enfermedad (230).
Se ha propuesto el uso de marcadores tumorales en orina como herramienta diagnóstica en pacientes que consultan por hematuria, como indicadores pronósticos de recurrencia y sobrevida de la enfermedad, y como detectores tempranos de enfermedad recurrente en los pacientes controlados. Entre las aplicaciones potenciales de pruebas de marcadores tumorales en orina para la vigilancia de pacientes se encuentran las pruebas seriadas para la detección temprana de la enfermedad recurrente, coadyuvantes de citología urinaria para mejorar la detección de recurrencia de la enfermedad, alternativas menos costosas y más objetivas que la citología urinaria, e indicadores para guiar la frecuencia de la evaluación cistoscópica en el seguimiento de pacientes con cáncer de vejiga. Para elaborar estas guías, se revisó la literatura relevante para el uso de marcadores tumorales en el cáncer de vejiga.
Se prestó especial atención a las revisiones, incluyendo las revisiones sistemáticas, pruebas prospectivas al azar que incluían el uso de marcadores, y guías emitidas por paneles de expertos. Cuando fue posible, las recomendaciones consensuadas del panel de la NACB se basaron en evidencias disponibles (es decir, fueron basadas en la evidencia).
Marcadores tumorales para cáncer de vejiga disponibles actualmente
En la Tabla III se listan los marcadores tumorales de cáncer de vejiga que se encuentran disponibles actualmente y aquellos que están en desarrollo, junto con la evaluación de cada marcador y el nivel de evidencia (LOE) para su uso clínico. Los sistemas de clasificación LOE (58) y SOR (fuerza de la recomendación) (231) se han aplicado tal como se lo describió anteriormente (2) [SOR (231), A=alta (investigaciones posteriores muy probablemente no puedan cambiar la confianza del panel en la estimación del efecto); B=moderada (investigaciones posteriores probablemente tengan un impacto importante en la confianza del panel en la estimación del efecto y es probable que cambie la estimación); C=baja (investigaciones posteriores muy probablemente tengan un efecto importante en la confianza del panel en la estimación del efecto y es probable que cambien la estimación); D= muy baja (cualquier estimación de efecto es muy incierta)]. Tal como se indicó en la Tabla III, la US Food and Drug Administration (FDA) ha aprobado seis pruebas de marcadores tumorales para su uso en la atención de rutina de los pacientes, todas las cuales se miden en orina.
Tabla III. Marcadores urinarios para cáncer de vejiga útiles y potencialmente útiles
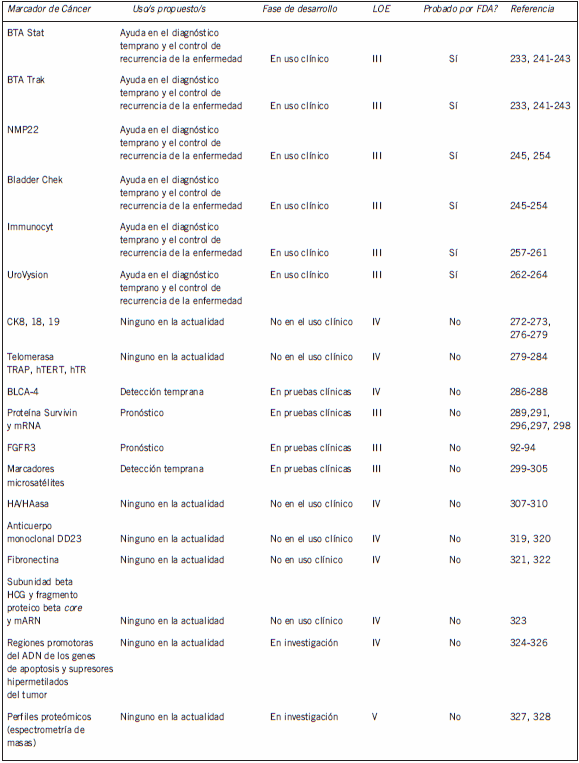
Marcadores tumorales en orina en cáncer de vejiga: recomendaciones de la NACB
En la actualidad, y de acuerdo con las guías de práctica de la NCCN para el cáncer de vejiga (232), no se puede recomendar ningún marcador tumoral para uso en el diagnóstico de rutina y en manejo clínico del cáncer de vejiga. Entre ellos se encuentran las pruebas para la realización de diagnóstico diferencial, la evaluación del pronóstico, la estadificación de la enfermedad, y el control de los pacientes para la detección temprana de una enfermedad recurrente.
No existe ningún dato de prueba clínica prospectiva que establezca la utilidad de alguno de los marcadores aprobados por la FDA o los marcadores propuestos para incrementar el tiempo de sobrevida, disminuir el costo del tratamiento, o mejorar la calidad de vida de los pacientes con cáncer de vejiga. En el siguiente informe, se describirán los marcadores aprobados por la FDA y la variedad de marcadores recientemente propuestos.
MARCADORES APROBADOS POR LA FDA PARA EL CÁNCER DE VEJIGA
Pruebas BTA-STAT y TRAK para Factor H del complemento y proteínas relacionadas
La prueba BTA-Stat (Polymedco, Cortlandt Manor, NY) detecta el factor de complemento H (CFH) y proteínas relacionadas con CFH en orina (233). El Factor H, una proteína 155-kDa, tiene un papel central en la regulación del camino alternativo de activación del complemento para la prevención del daño a las células saludables mediado por el complemento. Se han identificado al menos otras cuatro proteínas relacionadas con el factor H como productos de un cluster de genes en el cromosoma 1 llamados reguladores de la región de activación del complemento, y a pesar de que algunas de estas proteínas tienen actividad regulatoria complementaria, otras no la tienen (233).
La prueba BTA-Stat permite la detección semicuantitativa de proteínas antigénicas CFH y relacionados con la CFH por medio del uso de un doble anticuerpo monoclonal mediante un dispositivo inmunocromatográfico point-of-care. Se informa en varias oportunidades que la prueba BTA-Stat tiene sensibilidades dentro del rango 50%-83% (234-238) y especificidades dentro del rango 60%-92% (236) (239) (240), tanto para tumores no-invasivos (Tis, Ta, T1) como invasivos (T2-T4). Se informa que los resultados falso-positivos ocurren en algunos pacientes luego del trauma y en pacientes con infección de la vejiga o del tracto urinario, con nefritis, cálculos urinarios o hiperplasia prostática benigna (241).
La prueba BTA-Trak es una versión de inmunoensayo enzimático cuantitativo de la prueba BTA-Stat. El fabricante informa sensibilidades de 67% (Tis), 59% (Ta), 92% (T1), y 89% (T2-T4) para los estadios de cáncer de vejiga indicados. Se observan especificidades del 60% en enfermedad renal benigna, infecciones del tracto urinario y enfermedades de transmisión sexual, y aumentan 80%-90% en otras enfermedades genitourinarias distintas.
Ambas pruebas tienen sensibilidades comparables con la de la citología para tumores de grado alto y mejores que la citología para los tumores de grado bajo. Sin embargo, debido a su alta tasa falso-positiva, estas pruebas no son lo suficientemente precisas como para ser usadas para el screening o la detección temprana de los tumores de vejiga. El panel de la NACB por lo tanto no recomienda las pruebas BTA-Stat o Trak para su uso en el screening o el diagnóstico.


PROTEÍNA DE LA MATRIZ NUCLEAR
La prueba de la proteína de la matriz nuclear 22 (NMP22) (Matritech, Drummondville, Quebec, Canadá) es una prueba de anticuerpos monoclonales doble diseñada para medir cuantitativamente la proteína del aparato mitótico. Este componente de la matriz nuclear es sobreexpresado en el cáncer de vejiga y se elimina por la orina en cantidades mayores. NMP22 no es estable en la orina, y se recomienda el uso de un preservante de proteínas (228). Los datos que surgen de pruebas clínicas demostraron que cuando la prueba NMP22 se realizaba entre 6 y 40 días posteriores a la cirugía predecían de manera correcta la presencia de la enfermedad recurrente en la primera citoscopía de seguimiento en el 71% de los pacientes (24 de 34) con resultados NMP22 positivos (245). En los pacientes con valores de prueba NMP22 negativos, 86% (61 de 71) no tuvieron evidencia clínica de la enfermedad en la primera cistoscopía de seguimiento. Miyanaga et al (246) informaron resultados similares para la prueba NMP22 pero con 35% de tasa falso-positiva. En ese estudio y en un informe de seguimiento (247), NMP22 tuvo claramente un mejor desempeño que la citología de orina para la detección de cáncer de vejiga. También Stampfer et al y otros investigadores informaron resultados similares en un estudio multicéntrico que incluyó 171 pacientes con 274 citoscopías (249) (250).
Se encuentra disponible una versión point-of-care de la prueba NMP22 llamada Bladder Chek NMP22 (251). Un informe mencionó el efecto falso-positivo de los eritrocitos en esta prueba (252), mientras que otro informe reciente sugirió que los resultados falso-positivos de NMP22 eran atribuibles a la presencia de leucocitos (253). En una comparación reciente de Bladder Chek con la citología, en la que se testearon 1.331 pacientes con hematuria, la prueba Bladder Chek tuvo una sensibilidad de 55,7%, mientras que la citología detectó 15,8% de los cánceres. La especificidad del Bladder Chek fue de 85,7%, en comparación con el 99,2% de especificidad para citología de orina (254). La alta tasa falsopositiva de las pruebas basadas en NMP22 ha limitado su aceptación general para el uso de rutina en la atención del paciente.
Los valores informados para la sensibilidad de la prueba ELISA NMP22 se ubican entre 47% y 100% (255). Otros estudios han demostrado que la NMP22 tiene un peor desempeño en la vigilancia que en la detección del cáncer de vejiga, a pesar de que NMP22 tiene una mejor sensibilidad para la vigilancia que la citología (256). Se informó que la combinación de NMP22 y cistoscopía era más sensible que la cistoscopía sola para detectar recurrencias (222). No obstante, se evaluó la NMP22 como un complemennto de la cistoscopía o de la citología solas (256). En conclusión, la prueba de NMP22 es fácil de realizar, tiene mejor sensibilidad que la citología y una especificidad razonable, y asimismo es sensible en los tumores de bajo grado (247) (249) (250). A pesar de que la tasa falso-positiva es alta, la NMP22 puede ser superior que la citología en sensibilidad, y al seleccionar al paciente cuidadosamente, se podría mejorar la especificidad de NMP22.
La FDA ha aprobado la prueba NMP22 para su uso como herramienta de diagnóstico en pacientes en riesgo de cáncer de vejiga o con síntomas de tenerlo (255).

Prueba ImmunoCyt
La prueba ImmunoCyt (Diagno-Cure, Quebec, Canadá) detecta los marcadores asociados al cáncer de vejiga presentes en células exfoliadas utilizando un cocktail de anticuerpos fluorescentes (19A211, M344, y LDQ10; (257)). El anticuerpo monoclonal 19A211 detecta antígeno carcinoembrionario de alto peso molecular, mientras que M344 y LDQ10 detectan una mucina relacionada con el cáncer. Según un informe reciente, la prueba tiene una sensibilidad del 81% y una especificidad del 75% en la detección del cáncer de vejiga (258). La prueba ImmunoCyt se evaluó en varias investigaciones previas (259) (260) con hallazgos similares (259) (260). Cuando se la utiliza con citología, la prueba ImmunoCyt parece mejorar la detección de tumores de bajo grado (261).
Prueba UroVysion
Multitarget FISH detecta las células cancerígenas sobre la base de la aneuploidía de cromosomas seleccionados. La prueba UroVysion (Vysion) emplea sondas del centrómero específicas de los cromosomas 3, 7, y 17, una sonda específica para el sitio 9p21 para detectar aneuploidía asociada con cáncer de vejiga (262). Un estudio multicéntrico de la prueba UroVysion demostró un 71% de sensibilidad y 94,5% de especificidad para el cáncer de vejiga, que es mucho mejor que el de la prueba BTA Stat (263). Friedrich et al informaron un hallazgo similar en una comparación entre UroVysion con BTA Stat y NMP22 (264).
En otros estudios, la sensibilidad de la prueba UroVysion se sitúa entre 69% y 87% (255)(265-267). Esta prueba tiene una excelente sensibilidad para detectar un carcinoma in situ y tumores de alto grado/alto estadio (rango 83% a 100%). De hecho, el análisis FISH puede ser de utilidad para predecir la enfermedad oculta en aquellos pacientes sin ninguna evidencia cistoscópica de tumor y, por lo tanto, resolver casos con citología ambigua, y para monitorear la respuesta a la terapia.
Un estudio demostró que el 89% de los pacientes con resultados negativos de biopsia de vejiga y citología atípica en el entorno de FISH positivo desarrollaban carcinoma de células transicionales probado por biopsia, dentro de los siguientes 12 meses (268). Los resultados de estudios recientes sugieren que diferentes marcadores en la prueba UroVysion pueden tener distinta significancia cuando se los utiliza para predecir el comportamiento biológico del cáncer de vejiga (269). Varios estudios han demostrado que UroVysion también puede ser útil para monitorear a los pacientes luego del tratamiento con Bacilo Calmette-Guerin (270) (271).
Por lo tanto, la prueba UroVysion parece ser una prueba promisoria para la detección de cáncer de vejiga de alto grado, al tiempo que parece tener un potencial para predecir la recurrencia del cáncer y su progresión dentro de los 6 a 12 meses. En la actualidad, la realización de pruebas FISH debería reservarse para situaciones clínicas seleccionadas, en las que pueden brindar más información que la citología. El alto costo y la complejidad de la prueba-que requiere de personal muy capacitado y de un equipamiento sofisticado-han retrasado su adopción como práctica de rutina. Entre las limitaciones que tiene se puede mencionar que requiere de células uroteliales intactas y que no hay consenso acerca de qué constituye un resultado positivo (228).

BIOMARCADORES PROPUESTOS NO APROBADOS POR LA FDA
Citoqueratinas
Las citoqueratinas (CQ) son características de los filamentos intermedios de las células epiteliales. Se produce una sobre-expresión de ciertas citoqueratinas en el carcinoma de células transicionales de la vejiga (272). En estudios recientes que utilizaron un método ELISA para medir el fragmento citoqueratina 19 (CYFRA 21- 1) se demostró 75% a 97% de sensibilidad y aproximadamente 70% de especificidad (255). Un ensayo específico para CQ19 urinaria (CYFRA 21-1) también ha demostrado tener una alta sensibilidad y especificidad para el cáncer de vejiga (273). Sin embargo, el desempeño de este marcador no es bueno en el cáncer de vejiga en estadio temprano, quizás porque refleje el hecho de que las concentraciones de CYFRA 21-1 están influidas por enfermedades urológicas benignas e instilaciones intravesicales (274). Se han medido las concentraciones de CQ20 en células exfoliadas utilizando tanto RT-PCR como técnicas inmunocitoquímicas (255)(275). La sensibilidad de CQ20 que detecta cada método varía entre 78% y 87%, con especificidad entre 55% y 80% (255)(275).
La prueba del antígeno polipeptídico tisular (TPA) (Sangtec Medical, Stockholm, Sweden) emplea antisueros policlonales para la detección de CQ8, 18 y 19. A pesar de que se informa que la sensibilidad general es de 80%, una tasa faso-positiva de 30%-40% ha limitado el uso de TPA en la atención de rutina del paciente (276). Posteriormente, se desarrolló una prueba específica de polipéptido tisular (TPS) (IDL Biotech, Bromma, Sweden), la que emplea anticuerpos monoclonales contra CQ8 y 18 (277). Otra versión, llamada prueba urinaria de cáncer de vejiga (UBC) (IDL) también detecta CQ8 y 18. Un informe preliminar sugiere una sensibilidad de 65% y una especificidad del 92% para esta prueba (276) (278). En un estudio de comparación de método, la prueba UBC tuvo un mejor rendimiento que las pruebas BTA Stat y NMP22, demostrando una mayor sensibilidad y especificidad para cáncer de vejiga (279).
No obstante, en general la relativamente baja especificidad de los marcadores de citoqueratina, particularmente referidos a los pacientes con condiciones inflamatorias benignas, limita su aplicabilidad clínica.
Telomerasa
Los telómeros son regiones ubicadas en el extremo de los cromosomas humanos y están compuestas por muchas secuencias repetitivas cortas e idénticas de TTAGGG. Su función es la de estabilizar y proteger a los cromosomas (279) (280). Con cada ciclo celular, los extremos de los telómeros se acortan, hasta que alcanzan una longitud crítica, luego de la cual la división celular origina una ruptura en el telómero. La telomerasa es una enzima ribonucleoproteíca que agrega repeticiones de telómero para mantener la longitud del mismo. La telomerasa está inactivada en el tejido epitelial humano normal, pero se reactiva en la neoplasia (279). La telomerasa tiene dos componentes principales, un molde de ARN y una subunidad enzimática.
El ensayo Telomeric Repeat Amplification Protocol (TRAP) (Geron, Menlo Park, CA) mide la actividad enzimática de la telomerasa. Las repeticiones teloméricas se sintetizan in vitro y se amplifican por PCR, y los productos se visualizan por distintos métodos (279). En un estudio tisular de tumores de vejiga, 86% de las muestras (48 de 56) demostraron ser positivas para telomerasa, pero no se detectó ninguna actividad en el tejido de vejiga no-neoplásico.
El mismo estudio evaluó las células exfoliadas de 109 muestras de orina de pacientes urológicos, 26 de los cuales tenían cáncer de vejiga. Los autores informaron 62% de sensibilidad y 96% de especificidad para la actividad telomerasa en células uroteliales exfoliadas (280). Entro los avances que se han realizado en la medición de la telomerasa se encuentran los ensayos RT-PCR para el ARN de la telomerasa humana (hTR) y ARNm de la transcriptasa inversa de la telomerasa humana (hTERT). Estos ensayos han demostrado una sensibilidad de 83% para hTR y de 80% para hTERT (281) (282). Sanchini et al compararon los ensayos TRAP y hTERT y confirmaron la alta sensibilidad de ambos ensayos para telomerasa, pero sugirieron que el ensayo hTERT puede estar sujeto a una alta tasa de falso-positivos en pacientes con inflamación del tracto urinario (283). Saad et al informaron que el uso combinado del ensayo TRAP con NMP22 ofrecía sensibilidad y especificidad comparable con la citología de orina (284). No obstante, muchos pacientes con cáncer de vejiga tienen otras comorbilidades, lo cual limita la aplicabilidad de los ensayos de telomerasa. En un estudio, la sensibilidad fue tan baja como 7% debido a la inactivación de la enzima de telomerasa en la orina (285). En conclusión, los ensayos de telomerasa no son útiles en su forma actual para detectar y controlar el cáncer de vejiga.
BLCA-4
Se ha descripto una proteína de matriz nuclear específica para el cáncer de vejiga (BLCA-4) (286) (287). Se identificaron las proteínas BLCA en geles bidimensionales y se las secuenció; posteriormente aumentaron los anticuerpos contra los péptidos sintéticos correspondientes con esas secuencias. Los datos preliminares del inmunoensayo mostraron la proteína BLCA-4 presente en la orina de 53 de los 54 pacientes de cáncer de vejiga (4 en estadio Tis, 25 en estadío Ta-T1, 13 en estadio T2-T3, y 6 en estadio T4). Las concentraciones de BLCA-4 en orina en el total de los 51 controles sanos estuvieron por debajo del límite superior del intervalo de referencia. Sin embargo, 38 de los 202 pacientes con lesión en la médula espinal tuvieron valores elevados. Se halló posteriormente un tumor superficial en solamente uno de estos 38 pacientes (288). Debido a que los pacientes con lesiones en la médula espinal se encuentran en mayor riesgo de desarrollar un cáncer de vejiga, estos pacientes requerirán un seguimiento adicional para evaluar el papel diagnóstico de BLCA-4. Hay estudios clínicos en proceso para confirmar datos preliminares alentadores acerca de la utilidad de BLCA-4 en el cáncer de vejiga.
Survivina
La proteína survivina es un inhibidor de la apoptosis que se encuentra asociada con el huso mitótico (289) y se expresa en los cánceres más comunes (290), con baja expresión en tejidos adultos normales pero alta en tejidos cancerígenos y líneas celulares transformadas (291). La expresión de survivina puede detectarse en todos los tejidos cancerígenos de la vejiga, pero no en especímenes del urotelio normales (292) (293). Los patrones de expresión de survivina en pacientes con cáncer de vejiga se pueden examinar en orina, por RT-PCR de ARNm de la survivina (294) (295). Smith et al han desarrollado un inmunoensayo semicuantitativo policlonal para evaluar el papel de la survinina como marcador urinario para cáncer de vejiga (291). La proteína se detectó en todos los 46 casos nuevos y recurrentes de cáncer de vejiga, pero en ninguno de los 17 individuos sanos. La survivina estuvo presente en 3 de los 35 pacientes que habían sido previamente tratados por cáncer de vejiga pero que habían tenido evaluaciones cistoscópicas negativas (291). Más recientemente, Shariat et al informaron sensibilidad y especificidad y valores predictivos positivos y negativos para la proteína survivina de 64%, 93%, 92%, y 67%, respectivamente, en muestras de orina precistoscópicas (296). En este estudio, la survivina en orina mejoró el desempeño de la prueba NMP22 para detectar el cáncer de vejiga. La detección de transcripciones de ARNm de survivina en células exfoliadas y lavados vesicales, en lugar de la proteína survivina, pueden mejorar aun más la detección de cáncer de vejiga (297).
En un estudio, la detección de ARNm de survivina en sedimento urinario por medio de RT-PCR mostró alta sensibilidad (94%) y especificidad (95%) para el cáncer de vejiga y puede resultar ser útil para el screening y el control de rutina de los pacientes (292). De modo similar, Schultz et al identificaron survivina como el candidato más promisorio para distinguir entre los pacientes con carcinoma celular urotelial Ta primario y un intervalo libre de recurrencias largo (71,4%) o corto (69,6%) (298).
En el futuro, el análisis de la expresión de ARNm de survivina puede ayudar al urólogo a individualizar el tratamiento del paciente y prevenir cistoscopías innecesarias en un subgrupo de pacientes con cáncer de vejiga.
Detección de microsatélites
A lo largo de todo el genoma se encuentran presenten secuencias repetitivas de ADN, cada una de ellas con uno a cuatro pares de bases, que pueden sufrir cambios por mutaciones asociadas con la neoplasia, por lo cual sirven de marcadores genéticos del cáncer. El cambio genético más común que se ve en el cáncer de vejiga es la pérdida de heterozigosidad en el cromosoma 9.
Entre el 60% y 70% de los neoplasmas de vejiga muestran pérdida de heterozigosidad tanto en el brazo largo como en el corto del cromosoma 9, lo cual indica que la pérdida de los genes supresores puede ser el episodio temprano iniciador de carcinogénesis de vejiga (299) (300). Por medio del uso de 20 microsatélites marcadores ADN , Mao et al (301) detectaron 95% de los pacientes con cáncer de vejiga. Steiner et al (302) testearon dos marcadores de microsatélites en muestras de orina seriadas de 21 pacientes que habían sido tratados por cáncer de vejiga. Se detectaron lesiones recurrentes en 10 de los 11 pacientes en los que se había verificado independientemente que tenían la enfermedad recurrente. Los resultados de otros estudios distintos (303-305) que usaron diferentes paneles de marcadores de ADN sugieren que se podría identificar un pequeño conjunto de microsatélites marcadores que reflejen las alteraciones clave de ADN específicas y sensibles al cáncer de vejiga. Todos estos informes sugieren que el análisis de microsatélites de células exfoliadas es potencialmente útil para detectar cáncer de vejiga.
Se ha completado un estudio de validación prospectivo de centros múltiples para la detección de cáncer de vejiga incidental y predicción de la recurrencia. Este estudio fue iniciado por investigadores de la Johns Hopkins University y contó con el apoyo del National Cancer Institute Early Detection Research Network y sus resultados están pendientes. Un estudio similar conducido en Holanda para la detección y seguimiento de la enfermedad en un grado bajo, que calculó el valor de los polimorfismos de microsatélites para la detección del cáncer de vejiga demostró una sensibilidad de 58% y una especificidad de 73% para la detección de la recurrencia (306). Una prueba que resultó positiva de manera persistente estuvo asociada con un 83% de probabilidad de recurrencia a los 2 años.
Ácido hialurónico y hialuronidasa
El ácido hialurónico (HA), el ligando glicosaminoglicano para CD44, puede promover la adhesión, migración y angiogénesis de células tumorales.
La hialuronidasa (HAasa) degrada HA en fragmentos activos angiogénicamente. Lokeshwar et al (307) han demostrado que la prueba HA tiene una sensibilidad de 83% y una especificidad de 90% para detectar cáncer de vejiga. Además, hallaron la HAasa elevada de 5 a 8 veces en la orina de los pacientes con tumores grado 2 y 3, en comparación con los individuos sanos.
La medición de HAasa ha demostrado una sensibilidad del 100% y una especificidad del 89% para la detección de estos tumores de vejiga de grado alto en 139 pacientes (308). Hautmann y colaboradores han usado estos analitos juntos en una prueba HA-HAasa combinada (309). En dos estudios de comparación de métodos, la prueba de HA-HAasa mejoró la actuación de la prueba ImmunoCyt (309) y las BTA-Stat y UBC (310) en la detección del cáncer de vejiga.
Receptor 3 del factor de crecimiento de fibroblastos
Un avance reciente de importancia en el conocimiento de la patogénesis molecular del cáncer de vejiga ha sido la identificación de las mutaciones activadoras del receptor 3 del factor de crecimiento para fibroblastos (FGFR3) (311) (312). FGFR3 regula el crecimiento, la diferenciación y la angiogénesis celular (313). Las mutaciones de FGFR3 identificadas en el cáncer de vejiga son idénticas a aquellas presentes en los trastornos autosómicos dominantes del músculo esquelético (314). Se ha propuesto que las mutaciones FGFR3, que ocurren predominantemente en el tejido tumoral papilar no invasivo de la vejiga de bajo grado, están asociadas con la mejora en la sobrevida de los pacientes con tumores Ta y T1 (315).
Las mutaciones FGFR3 caracterizan la vía papilar del carcinoma de bajo grado de vejiga y la frecuencia de la mutación disminuye de manera constante entre los tumores no-invasivos a medida que el estadio y el grado aumentan. La presencia de mutaciones FGFR3 podría ser una variable para el pronóstico (316). Sin embargo, ningún estudio hasta la fecha ha demostrado si la mutación FGFR3 tiene independencia pronóstica significativa (317). La detección de la mutación FGFR3 puede en el fututo proporcionar una herramienta útil en el manejo estándar de los pacientes con tumores papilares de vejiga de bajo grado (228) (316) (318). El panel de la NACB recomienda que esto se estudie de manera más exhaustiva en pruebas clínicas prospectivas.
Otros marcadores propuestos
El anticuerpo monoclonal DD23 reconoce un antígeno 185-kDa expresado por células de cáncer de vejiga y se ha propuesto como suplementario a la citología para la detección de cáncer de vejiga (319) (320).
La fibronectina urinaria (321) (322) y la subunidad β de la gonadotrofina coriónica (HCG) y el fragmento β core (proteína y el ARNm trasncripto) también pueden ser marcadores para el carcinoma celular transicional de la vejiga (323). La detección de la hipermetilación de las regiones promotoras de los genes supresores de tumores y de los genes de la apoptosis parece tener un valor diagnóstico potencial para el cáncer de vejiga (324-326).
Recientemente, el uso de perfiles proteómicos de orina se ha sugerido como abordaje diagnóstico para el cáncer de vejiga (327) (328).
Papel de los marcadores de orina en la detección temprana del cáncer de vejiga
Casi todos los casos de cáncer de vejiga se descubren durante el examen médico de los pacientes que consultan por hematuria (329), pero la mayoría de los casos de hematuria no están causados por cáncer de vejiga. La enfermedad urológica se detecta en el 50% de los pacientes que consultan por hematuria (en los cuales la anomalía más común es la hipertrofia prostática benigna), y el cáncer de vejiga se detecta en 10% de los pacientes con hematuria evidente y 2%-3% de los pacientes con microhematuria (330-332). El chequeo de los pacientes con hematuria es costoso y puede requerir citología, cistoscopía, urografía intravenosa o TC (333). Por lo tanto, los marcadores tumorales podrían ser útiles para identificar a los pacientes en este grupo de alto riesgo, que requiere más chequeo clínico intensivo para el cáncer de vejiga. Zippe et al informaron acerca del valor de la prueba de orina NMP22 para evaluar a 330 pacientes con hematuria (334). La prueba NMP22, usada con valor de corte de 10,0 U/mL detectó los 18 casos de cáncer de vejiga con 45 casos falso-positivos (sensibilidad, 100%; especificidad, 85%). En este estudio, se podrían haber evitado 267 cistoscopías innecesarias si la prueba NMP22 hubiera guiado la cistoscopía. En una prueba clínica presentada por la FDA (como datos de aprobación previos al mercado), los resultados de la prueba NMP22 fueron elevados en 69,6% de los 56 casos de cáncer de vejiga que se detectaron en el grupo de alto riesgo. En este informe, la especificidad fue de 67,7% (335). La prueba NMP22 ha sido aprobada por la FDA para uso como ayuda en el diagnóstico de cáncer de vejiga en los individuos con factores de riesgo alto o que tienen síntomas de cáncer de vejiga. Es muy probable que otros marcadores en orina (por ejemplo, BTA-Stat, UroVysion e Immunocyt) puedan tener también valor para la detección del cáncer en sujetos que consultan por hematuria. La alta tasa falso-positiva es la crítica más importante que reciben las pruebas basadas en la orina cuando se las usa para evaluar pacientes que consultan por hematuria o cuando se las usa en la vigilancia de los pacientes.
La baja tasa falso-negativa de estas pruebas es su fortaleza, lo cual las lleva a un alto valor predictivo negativo que descarta la enfermedad de manera eficaz en una proporción importante de pacientes, por lo que elimina controles clínicos innecesarios para el cáncer de vejiga. La alta tasa de falso-positivos de los biomarcadores urinarios ha limitado su papel como complementos de la cistoscopía y la citología para la detección de la enfermedad recurrente. Más importante aún, no existen datos basados en la evidencia que demuestren que la vigilancia basada en los biomarcadores urinarios lleve a mejores resultados en la supervivencia de los pacientes, a una mejor calidad de vida, o a menores gastos en la atención de la salud.
Papel de los marcadores tisulares para el pronóstico
Una importante cantidad de la investigación sigue estando dirigida a la identificación de marcadores que predicen el potencial agresivo de los tumores de vejiga no invasivos. Tal información puede dar lugar a protocolos de vigilancia más efectivos y permitir un tratamiento más agresivo en aquellos pacientes con tumores que más probablemente vayan a progresar a enfermedad invasiva o metastásica (336). Stein et al han desarrollado una revisión exhaustiva de una variedad de marcadores biológicos que se ha informado tienen valor pronóstico (336). Más recientemente, se ha informado que el p53 y otros genes de control del ciclo celular (337) (338), los transcriptos del gen HCG β (339), y distintas proteínas de adhesión y de la matriz celular y genes expresados diferencialmente (tumores en estadio temprano vs tumores en estadio tardío) tienen valor pronóstico (340). Sin embargo, en la actualidad, ninguno de estos marcadores se ha validado todavía para uso en la atención de rutina de los pacientes.
A pesar de que muchos estudios han demostrado que la prevalencia de las alteraciones de p53 en el cáncer de vejiga aumentan con el estadio y el grado (341) (342), no existe ninguna evidencia definitiva de que la sobreexpresión de p53 sea un factor pronóstico independiente (342).
Algunos resultados, sin embargo, sugieren que las mutaciones genéticas de la proteína tumoral p53 (TP53) pueden ser factores pronósticos independientes para una pobre sobrevida libre de progresión en el cáncer de vejiga no invasivo (343-345). Además, las mutaciones en ciertos sitios del gen TP53, particularmente en el exón 8, pueden ser responsables de un peor pronóstico debido a que estos sitios implican la función biológica de p53 (346). Las mutaciones en dominios funcionales y estructurales definidos de p53 pueden por lo tanto servir de marcadores biológicos moleculares útiles para determinar las estrategias de pronóstico y tratamiento en los pacientes, con carcinomas celulares transicionales no invasivos. Este hallazgo es potencialmente aun más significativo debido a que las mutaciones del TP53 se pueden analizar en células urinarias por medio de métodos no invasivos (347)(348).
En la medida en que estén disponibles técnicas más novedosas y rápidas para análisis genéticos, tales pruebas se harán de rutina en el futuro.
Otro fuerte indicador de la progresión del tumor en los pacientes de cáncer de vejiga ha demostrado ser también la hipermetilación del gen del factor 1 modulado por poliamida (PMF1) (349). Además, se ha informado que la pérdida de la expresión de la proteína PMF1 estratifica los tumores de vejiga histopatológicamente y predice el resultado clínico (349) (350).
Papel de los marcadores urinarios para la vigilancia de los pacientes
Muchos estudios han establecido el valor de las pruebas de marcadores tumorales en orina para la detección temprana de tumores de vejiga recurrentes, pero hasta el momento, estas pruebas no han podido reemplazar a la cistoscopía y a la citología de rutina en el manejo de los pacientes con cáncer de vejiga. Por el contrario, estos marcadores pueden usarse como anexos complementarios que dirijan el uso más efectivo de los procedimientos clínicos, por lo que potencialmente se reducirían los costos de la vigilancia de los pacientes.
Los pacientes con lesiones superficiales de grado bajo (Ta, grado 1 y 2) se encuentran en menor riesgo de recurrencia que los pacientes con tumores Ta de grado III y tumores T1; y estos pacientes en menor riesgo probablemente necesiten un seguimiento menos intensivo (248).
Los marcadores urinarios que se usan en la vigilancia de los pacientes han sido criticados alguna vez por su baja sensibilidad para detectar la enfermedad (351) (352), pero en la mayoría de los estudios han mejorado de manera significativa la detección del cáncer de vejiga cuando se los utiliza en conjunción con la citología y la cistoscopía. Debido a su baja sensibilidad, la citología de orina tiene sus limitaciones para detectar el carcinoma in situ (Tis) y los tumores de vejiga de bajo grado (353). Parece que los marcadores urinarios pueden ayudar en la detección temprana de la recurrencia en pacientes con carcinoma in situ y tumores superficiales de bajo grado (354).
Puntos clave: Marcadores tumorales en cáncer de vejiga
La disponibilidad de muchos marcadores nuevos para cáncer de vejiga ofrece la posibilidad de mejorar la tasa de detección de cáncer al combinar el uso de los marcadores seleccionados, medidos simultáneamente o bien secuencialmente (355). El objetivo de la realización de tal panel de pruebas deberá ser la mejora tanto de la sensibilidad como de la especificidad para la detección del cáncer de vejiga. Sin dudas, son necesarias las pruebas clínicas prospectivas para probar el valor de tales paneles antes de que puedan implementarse en la atención de rutina del paciente (356). También debe notarse que la estabilidad de estos marcadores tumorales debe definirse mejor para minimizar los resultados falso-negativos de las pruebas. Se podría lograr un uso más efectivo de estas pruebas para la detección del cáncer (357) si se mejoraran las definiciones de las condiciones de esta enfermedad que puedan producir resultados falso-positivos para los marcadores en orina.
1. Field M, Lorh K, eds. Clinical practice guidelines: directions for a new program. Washington, DC: National Academy Press,1990, pp 168. [ Links ]
2. Diamandis EP, Hoffman BR, Sturgeon CM. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for the use of tumor markers. Clin Chem 2008; 54:1935-9. [ Links ]
3. Sturgeon CM, Hoffman BR, Chan DW, Ch'ng SL, Hammond E, Hayes DF, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in clinical practice: Quality requirements. Clin Chem 2008; 54: e1-10. [ Links ]
4. Jelic S. Hepatocellular carcinoma: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol 2009; 20: iv41-5. [ Links ]
5. Stuart KE, Stadler ZK. Hepatic carcinoma, primary. http://emedicine.medscape.com/article/282814-overview. [ Links ]
6. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet 2003; 362: 1907-17. [ Links ]
7. Bosch FX, Ribes J, Borràs J. Epidemiology of primary liver cancer. Semin Liver Dis 1999; 19: 271-85. [ Links ]
8. Tanaka Y, Hanada K, Mizokami M, Yeo AE, Shih JW, Gojobori T, Alter HJ. Inaugural Article: A comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci 2002; 99: 15584-9. [ Links ]
9. El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med 2003;139: 817-23. [ Links ]
10. Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, et al. Morbidity and mortality in compensated cirrosis type C: A retrospective follow-up study of 384 patients. Gastroenterology 1997; 112: 463-72. [ Links ]
11. Colombo.M, Berr F, Bruix J, Hauss J, Wands J, Wittekind C. In: Berr F, Bruix J, Hauss J, Wittekind C, Wands J, eds. Risk groups and preventive strategies. Malignant liver tumors: Basic concepts and clinical management (Falk Symposium). Kluwer Academic Publishers BV and Falk Foundation, 2003, pp 67-74. [ Links ]
12. Liaw YF, Tai DI, Chu CM, Lin DY, Sheen IS, Chen TJ, Pao CC. Early detection of hepatocellular carcinoma in patients with chronic type B hepatitis. A prospective study. Gastroenterology 1986; 90: 263-7. [ Links ]
13. Sun Z, Lu P, Gail MH, Pee D, Zhang Q, Ming L, et al. Increased risk of hepatocellular carcinoma in male hepatitis B surface antigen carriers with chronic hepatitis who have detectable urinary aflatoxin metabolite M1. Hepatology 1999; 30: 379-83. [ Links ]
14. Bruno S, Silini E, Crosignani A, Borzio F, Leandro G, Bono F, et al. Hepatitis C virus genotypes and risk of hepatocellular carcinoma in cirrhosis: A prospective study. Hepatology 1997; 25: 754-8. [ Links ]
15. Bruix J, Barrera JM, Calvet X, Ercilla G, Costa J, Sanchez- Tapias JM, et al. Prevalence of antibodies to hepatitis C virus in Spanish patients with hepatocellular carcinoma and hepatic cirrhosis. Lancet 1989; 2: 1004-6. [ Links ]
16. Colombo M, de Franchis R, Del Ninno E, Sangiovanni A, De Fazio C, Tommasini M, et al. Hepatocellular carcinoma in Italian patients with cirrhosis. N Engl J Med 1991; 325: 675-80. [ Links ]
17. Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993; 328: 1797-801. [ Links ]
18. Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med 1985; 313: 1256-62. [ Links ]
19. Zhou XD, Tang ZY, Yang BH, Lin ZY, Ma ZC, Ye SL, et al. Experience of 1000 patients who underwent hepatectomy for small hepatocellular carcinoma. Cancer 2001; 91: 1479-86. [ Links ]
20. Fattovich G, Giustina G, Schalm SW, Hadziyannis S, Sanchez- Tapias J, Almasio P, et al. Occurrence of hepatocellular carcinoma and decompensation in western European patients with cirrosis type B. The EUROHEP Study Group on Hepatitis B Virus and Cirrhosis. Hepatology 1995; 21: 77-82. [ Links ]
21. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet 1981; 2: 1129-33. [ Links ]
22. WHO. Hepatitis C: global prevalence. Wkly Epidemiol Rec 1997: 341-4. [ Links ]
23. European Association for the Study of the Liver. EASL Clinical Practice Guidelines. Management of chronic hepatitis B. J Hepatol 2009; 50: 227-42. [ Links ]
24. Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology 2007; 45: 507-39. [ Links ]
25. Ghany MG, Strader DB, Thomas DL, Seeff LB, and American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: An update. Hepatology 2009; 49: 1335-74. [ Links ]
26. Ryder SD. Guidelines for the diagnosis and treatment of hepatocellular carcinoma (HCC) in adults. Gut 2003; 52: iii1-8 (suppl 3). [ Links ]
27. Gogel BM, Goldstein RM, Kuhn JA, McCarty TM, Donahoe A, Glastad K. Diagnostic evaluation of hepatocellular carcinoma in a cirrhotic liver. Oncology (Williston Park) 2000; 14: 15-20. [ Links ]
28. Larcos G, Sorokopud H, Berry G, Farrell GC. Sonographic screening for hepatocellular carcinoma in patients with chronic hepatitis or cirrhosis: an evaluation. Am J Roentgenol 1998; 171: 433-5. [ Links ]
29. Sarasin FP, Giostra E, Hadengue A. Cost-effectiveness of screening for detection of small hepatocellular carcinoma in western patients with Child-Pugh class A cirrhosis. Am J Med 1996; 101: 422-34. [ Links ]
30. Schwartz JM, Carithers RL. Clinical features, diagnosis and screening for primary hepatocellular carcinoma. V. 17.2 May 2009. http://www.uptodateonline.com/patients/content/topic.do? topicKey=~kxb1bGB8WXPmsH [ Links ]
31. Kew MC, Dos Santos HA, Sherlock S. Diagnosis of primary cancer of the liver. Br Med J 1971; 4: 408-11. [ Links ]
32. A new prognostic system for hepatocellular carcinoma: a retrospective study of 435 patients: the Cancer of the Liver Italian Program (CLIP) investigators. Hepatology 1998; 28: 751-5. [ Links ]
33. Sugano S, Miyoshi K, Suzuki T, Kawafune T, Kubota M. Intrahepatic arteriovenous shunting due to hepatocellular carcinoma and cirrhosis, and its change by transcatheter arterial embolization. Am J Gastroenterol 1994; 89: 184-8. [ Links ]
34. Tietge UJ, Schöfl C, Ocran KW, Wagner S, Böker KH, Brabant G, et al. Hepatoma with severe non-islet cell tumor hypoglycemia. Am J Gastroenterol 1998; 93: 997-1000. [ Links ]
35. Kew MC, Fisher JW. Serum erythropoietin concentrations in patients with hepatocellular carcinoma. Cancer 1986; 58: 2485-8. [ Links ]
36. Knill-Jones RP, Buckle RM, Parsons V, Calne RY, Williams R. Hypercalcemia and increased parathyroidhormone activity in a primary hepatoma. Studies before and after hepatic transplantation. N Engl J Med 1970; 282: 704-8. [ Links ]
37. Yen TC, Hwang SJ, Wang CC, Lee SD, Yeh SH. Hypercalcemia and parathyroid hormone-related protein in hepatocellular carcinoma. Liver 1993; 13: 311-5. [ Links ]
38. Gregory B, Ho VC. Cutaneous manifestations of gastrointestinal disorders. Part I. J Am Acad Dermatol 1992; 26: 153-66. [ Links ]
39. Gomaa AI, Khan SA, Leen EL, Waked I, Taylor-Robinson SD. Diagnosis of hepatocellular carcinoma. World J Gastroenterol 2009; 15: 1301-14. [ Links ]
40. Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology 2005; 42: 1208-36. [ Links ]
41. Durand F, Regimbeau JM, Belghiti J, Sauvanet A, Vilgrain V, Terris B, et al. Assessment of the benefits and risks of percutaneous biopsy before surgical resection of hepatocellular carcinoma. J Hepatol 2001; 35: 254-8. [ Links ]
42. Silva MA, Hegab B, Hyde C, Guo B, Buckels JA, Mirza DF. Needle track seeding following biopsy of liver lesions in the diagnosis of hepatocellular cancer: a systematic review and meta-analysis. Gut 2008; 57: 1592-6. [ Links ]
43. Takayama T, Makuuchi M, Hirohashi S, Sakamoto M, Yamamoto J, Shimada K, et al. Early hepatocellular carcinoma as an entity with a high rate of surgical cure. Hepatology 1998; 28: 241-6. [ Links ]
44. Kojiro M, Tabor E. The evolution of pathologic features of hepatocellular carcinoma. In Tabor E, ed. Viruses and liver cancer: perspectives in medical virology. Amsterdam, the Netherlands, Elsevier Science, 2002, pp. 113-22. [ Links ]
45. Llovet JM, Fuster J, Bruix J. Intention-to-treat analysis of surgical treatment for early hepatocellular carcinoma: resection versus transplantation. Hepatology 1999; 30: 1434-40. [ Links ]
46. The Liver Cancer Study Group of Japan. Predictive factors for long term prognosis after partial hepatectomy for patients with hepatocellular carcinoma in Japan. Cancer 1994; 74: 2272-80. [ Links ]
47. Llovet JM, Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 2003; 37: 429- 42. [ Links ]
48. Lopez PM, Villanueva A, Llovet JM. Systematic review: Evidence- based management of hepatocellular carcinomaóan updated analysis of randomized controlled trials. Aliment Pharmacol Ther 2006; 23: 1535-47. [ Links ]
49. El-Serag HB, Marrero JA, Rudolph L, Reddy KR. Diagnosis and treatment of hepatocellular carcinoma. Gastroenterology 2008; 134: 1752-63. [ Links ]
50. Okada S. Local ablation therapy for hepatocellular carcinoma. Semin Liver Dis 1999; 19: 323-8. [ Links ]
51. Livraghi T, Giorgio A, Marin G, Salmi A, de SI, Bolondi L, et al. Hepatocellular carcinoma and cirrhosis in 746 patients: Long-term results of percutaneous ethanol injection. Radiology 1995; 197: 101-8. [ Links ]
52. Lencioni RA, Allgaier HP, Cioni D, Olschewski M, Deibert P, Crocetti L, et al. Small hepatocellular carcinoma in cirrhosis: randomized comparison of radio-frequency termal ablation versus percutaneous ethanol injection. Radiology 2003; 228: 235-40. [ Links ]
53. Fong Y, Sun RL, Jarnagin W, Blumgart LH. An analysis of 412 cases of hepatocellular carcinoma at a Western center. Ann Surg 1999; 229: 790-9. [ Links ]
54. Cho YK, Kim JK, Kim MY, Rhim H, Han JK. Systematic review of randomized trials for hepatocellular carcinoma treated with percutaneous ablation therapies. Hepatology 2009; 49: 453-9. [ Links ]
55. Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008; 48: 1312-27. [ Links ]
56. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359: 378-90. [ Links ]
57. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia- Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009; 10: 25-34. [ Links ]
58. Hayes DF, Bast RC, Desch CE, Fritsche H Jr, Kemeny NE, Jessup JM, et al. Tumor marker utility grading system: A Framework to evaluate clinical utility of tumor markers. J Natl Cancer Inst 1996; 88: 1456-66. [ Links ]
59. Johnson PJ, Diamandis EP, Fritsche HA, Lilja H, Chan DW, Schwartz MK. Tumor markers in primary malignanciesof the liver. In: Diamandis EP, Fritche HA, Lilja H, Chan DW, Schwartz MK, eds. Tumor markers: physiology, pathobiology, technology and clinical applications. Washington, DC, AACC Press, 2002, pp. 269-79. [ Links ]
60. Mizejewski GJ. Alpha-fetoprotein structure and function: relevante to isoforms, epitopes, and conformational variants. Exp Biol Med (Maywood) 2001; 226: 377-408. [ Links ]
61. Christiansen M, Hogdall CK, Andersen JR, Norgaard-Pedersen B. Alpha-fetoprotein in plasma and serum of healthy adults: preanalytical, analytical and biological sources of variation and construction of age-dependent reference intervals. Scand J Clin Lab Invest 2001; 61: 205-15. [ Links ]
62. Yoshima H, Mizuochi T, Ishii M, Kobata A. Structure of the asparagine-linked sugar chains of alpha-fetoprotein purified from human ascites fluid. Cancer Res 1980; 40: 4276-81. [ Links ]
63. Yamashita K, Taketa K, Nishi S, Fukushima K, Ohkura T. Sugar chains of human cord serum alpha-fetoprotein: characteristics of N-linked sugar chains of glycoproteins produced in human liver and hepatocellular carcinomas. Cancer Res 1993; 53: 2970-5. [ Links ]
64. Mora J, Gascon N, Tabernero JM, Germa JR, Gonzalez F. Alphafetoprotein-concanavalin A binding as a marker to discriminate between germ cell tumours and liver diseases. Eur J Cancer 1995; 31A: 2239-42. [ Links ]
65. Saitoh S, Ikeda K, Koida I, Suzuki Y, Kobayashi M, Tsubota A, et al. Diagnosis of hepatocellular carcinoma by concanavalin A affinity electrophoresis of serum alpha-fetoprotein. Cancer 1995; 76: 1139-44. [ Links ]
66. Aoyagi Y, Suzuki Y, Igarashi K, Saitoh A, Oguro M, Yokota T, et al. The usefulness of simultaneous determinations of glucosaminylation and fucosylation indices of alpha-fetoprotein in the differential diagnosis of neoplastic diseases of the liver. Cancer 1991; 67: 2390-4. [ Links ]
67. Taketa K, Ichikawa E, Taga H, Hirai H. Antibody-affinity blotting, a sensitive technique for the detection of a-fetoprotein separated by lectin affinity electrophoresis in agarose gels. Electrophoresis 1985; 6: 492-7. [ Links ]
68. Taketa K. Alpha-fetoprotein: Reevaluation in hepatology. Hepatology 1990; 12: 1420-32. [ Links ]
69. Johnson PJ, Leung N, Cheng P, Welby C, Leung WT, Lau WY, et al. "Hepatoma-specific" alphafetoprotein may permit preclinical diagnosis of malignant change in patients with chronic liver disease. Br J Cancer 1997; 75: 236-40. [ Links ]
70. Fujii Y, Taketa K, Aoi T, Taga H, Hirai H. Increased serum levels of monosialo-alpha-fetoprotein in hepatocellular carcinoma and other malignancies. Tumor Biol 1993; 14: 319-24. [ Links ]
71. Poon TC, Mok TS, Chan AT, Chan CM, Leong V, Tsui SH, et al. Quantification and utility of monosialylated alpha-fetoprotein in the diagnosis of hepatocellular carcinoma with nondiagnostic serum total alpha-fetoprotein. Clin Chem 2002; 48: 1021-7. [ Links ]
72. Shimizu K, Katoh H, Yamashita F, Tanaka M, Tanikawa K, Taketa K, et al. Comparison of carbohydrate structures of serum alpha-fetoprotein by sequential glycosidase digestion and lectin affinity electrophoresis. Clin Chim Acta 1996; 254: 23-40. [ Links ]
73. Johnson PJ, Poon TC, Hjelm NM, Ho CS, Ho SK, Welby C, et al. Glycan composition of serum alpha-fetoprotein in patients with hepatocellular carcinoma and non-seminomatous germ cell tumour. Br J Cancer 1999; 81: 1188-95. [ Links ]
74. Taketa K, Endo Y, Sekiya C, Tanikawa K, Koji T, Taga H, et al. A collaborative study for the evaluation of lectin-reactive alpha-fetoproteins in early detection of hepatocellular carcinoma. Cancer Res 1993; 53: 5419-23. [ Links ]
75. Sato Y, Nakata K, Kato Y, Shima M, Ishii N, Koji T, et al. Early recognition of hepatocellular carcinoma based on altered profiles of alpha-fetoprotein. N Engl J Med 1993; 328: 1802-6. [ Links ]
76. Taketa K, Sekiya C, Namiki M, Akamatsu K, Ohta Y, Endo Y, Kosaka K. Lectin-reactive profiles of alpha-fetoprotein characterizing hepatocellular carcinoma and related conditions. Gastroenterology 1990; 99: 508-18. [ Links ]
77. Yamashita F, Tanaka M, Satomura S, Tanikawa K. Prognostic significance of Lens culinaris agglutinin A-reactive alpha-fetoprotein in small hepatocellular carcinomas. Gastroenterology 1996; 111: 996-1001. [ Links ]
78. Wang SS, Lu RH, Lee FY, Chao Y, Huang YS, Chen CC, Lee SD. Utility of lentil lectin affinity of alpha-fetoprotein in the diagnosis of hepatocellular carcinoma. J Hepatol 1996; 25: 166-71. [ Links ]
79. Kumada T, Nakano S, Takeda I, Kiriyama S, Sone Y, Hayashi K, et al. Clinical utility of Lens culinaris agglutinin- reactive alphafetoprotein in small hepatocellular carcinoma: Special reference to imaging diagnosis. J Hepatol 1999; 30: 125-30. [ Links ]
80. Oka H, Saito A, Ito K, Kumada T, Satomura S, Kasugai H, et al. Multicenter prospective analysis of newly diagnosed hepatocellular carcinoma with respect to the percentage of Lens culinaris agglutinin-reactive alpha-fetoprotein. J Gastroenterol Hepatol 2001; 16: 1378-83. [ Links ]
81. Song BC, Suh DJ, Yang SH, Lee HC, Chung YH, Sung KB, Lee YS. Lens culinaris agglutinin-reactive alpha-fetoprotein as a prognostic marker in patients with hepatocellular carcinoma undergoing transcatheter arterial chemoembolization. J Clin Gastroenterol 2002; 35: 398-402. [ Links ]
82. Leerapun A, Suravarapu SV, Bida JP, Clark RJ, Sanders EL, Mettler TA, et al. The utility of Lens culinaris agglutinin-reactive alpha-fetoprotein in the diagnosis of hepatocellular carcinoma: evaluation in a United States referral population. Clin Gastroenterol Hepatol 2007; 5: 394-402. [ Links ]
83. Sterling RK, Jeffers L, Gordon F, Venook AP, Reddy KR, Satomura S, et al. Utility of Lens culinaris agglutinin-reactive fraction of alpha-fetoprotein and des-gamma-carboxy prothrombin, alone or in combination, as biomarkers for hepatocellular carcinoma. Clin Gastroenterol Hepatol 2009; 7: 104-13. [ Links ]
84. Carr BI, Kanke F, Wise M, Satomura S. Clinical evaluation of lens culinaris agglutinin-reactive alpha-fetoprotein and des-gammacarboxy prothrombin in histologically proven hepatocellular carcinoma in the United States. Dig Dis Sci 2007; 52: 776-82. [ Links ]
85. Tateishi R, Shiina S, Yoshida H, Teratani T, Obi S, Yamashiki N, et al. Prediction of recurrence of hepatocellular carcinoma after curative ablation using three tumor markers. Hepatology 2006; 44: 1518-27. [ Links ]
86. Marrero JA, Feng Z, Wang Y, Nguyen MH, Befeler AS, Roberts LR, et al. Alpha-fetoprotein, des-gamma carboxyprothrombin, and lectin-bound alpha-fetoprotein in early hepatocellular carcinoma. Gastroenterology 2009; 137: 110-8. [ Links ]
87. Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst 2008; 100: 698-711. [ Links ]
88. Johnson JR, Williams G, Pazdur R. End points and United States Food and Drug Administration approval of oncology drugs. J Clin Oncol 2003;21:1404-11. [ Links ]
89. Collier J, Sherman M. Screening for hepatocellular carcinoma. Hepatology 1998; 27: 273-8. [ Links ]
90. Chen DS, Sung JL, Sheu JC, Lai MY, How SW, Hsu HC, et al. Serum alpha-fetoprotein in the early stage of human hepatocellular carcinoma. Gastroenterology 1984; 86: 1404-9. [ Links ]
91. Kondo F, Wada K, Nagato Y, Nakajima T, Kondo Y, Hirooka N, et al. Biopsy diagnosis of well-differentiated hepatocellular carcinoma based on new morphologic criteria. Hepatology 1989; 9: 751-5. [ Links ]
92. Forner A, Vilana R, Ayuso C, Bianchi L, Sole M, Ayuso JR, et al. Diagnosis of hepatic nodules 20 mm or smaller in cirrhosis: prospective validation of the noninvasive diagnostic criteria for hepatocellular carcinoma. Hepatology 2008; 47: 97-104. [ Links ]
93. Yoshino M. Growth kinetics of hepatocellular carcinoma. Jpn J Clin Oncol 1983; 13: 45-52. [ Links ]
94. Barbara L, Benzi G, Gaiani S, Fusconi F, Zironi G, Siringo S, et al. Natural history of small untreated hepatocellular carcinoma in cirrhosis: a multivariate analysis of prognostic factors of tumor growth rate and patient survival. Hepatology 1992; 16: 132-7. [ Links ]
95. Okuda K. Early recognition of hepatocellular carcinoma. Hepatology 1986; 6: 729-38. [ Links ]
96. Forner A, Reig M, Bruix J. Alpha-fetoprotein for hepatocellular carcinoma diagnosis: the demise of a brilliant star. Gastroenterology 2009; 137: 26-9. [ Links ]
97. Lok AS, Sterling RK, Everhart JE, Wright EC, Hoefs JC, Di Bisceglie AM, et al. Des-gamma-carboxy Prothrombin and Alpha fetoprotein as Biomarkers for the Early Detection of Hepatocellular Carcinoma. Gastroenterology 2009; 138: 493-502. [ Links ]
98. Gupta S, Bent S, Kohlwes J. Test characteristics of alpha-fetoprotein for detecting hepatocellular carcinoma in patients with hepatitis C: A systematic review and critical analysis. Ann Intern Med 2003; 139: 46-50. [ Links ]
99. Trevisani F, D'Intino PE, Morselli-Labate AM, Mazzella G, Accogli E, Caraceni P, et al. Serum alpha-fetoprotein for diagnosis of hepatocellular carcinoma in patients with chronic liver disease: influence of HBsAg and anti-HCV status. J Hepatol 2001; 34: 570-5. [ Links ]
100. Tong MJ, Blatt LM, Kao VW. Surveillance for hepatocellular carcinoma in patients with chronic viral hepatitis in the United States of America. J Gastroenterol Hepatol 2001; 16: 553-9. [ Links ]
101. Cedrone A, Covino M, Caturelli E, Pompili M, Lorenzelli G, Villani MR, et al. Utility of alpha-fetoprotein (AFP) in the screening of patients with virus-related chronic liver disease: Does different viral etiology influence AFP levels in HCC? A study in 350 Western patients. Hepatogastroenterology 2000; 47: 1654-8. [ Links ]
102. Nguyen MH, Garcia RT, Simpson PW, Wright TL, Keeffe EB. Racial differences in effectiveness of alpha-fetoprotein for diagnosis of hepatocellular carcinoma in hepatitis C virus cirrhosis. Hepatology 2002; 36: 410-17. [ Links ]
103. Peng YC, Chan CS, Chen GH. The effectiveness of serum alpha-fetoprotein level in anti-HCV positive patients for screening hepatocellular carcinoma. Hepatogastroenterology 1999; 46: 3208-11. [ Links ]
104. Gebo KA, Chander G, Jenckes MW, Ghanem KG, Herlong HF, Torbenson MS, et al. Screening tests for hepatocellular carcinoma in patients with chronic hepatitis C: A systematic review. Hepatology 2002; 36: S84-S92. [ Links ]
105. Takano S, Yokosuka O, Imazeki F, Tagawa M, Omata M. Incidence of hepatocellular carcinoma in chronic hepatitis B and C: a prospective study of 251 patients. Hepatology 1995; 21: 650-5. [ Links ]
106. Sherman M, Peltekian KM, Lee C. Screening for hepatocellular carcinoma in chronic carriers of hepatitis B virus: Incidence and prevalence of hepatocellular carcinoma in a North American urban population. Hepatology 1995; 22: 432-8. [ Links ]
107. Pateron D, Ganne N, Trinchet JC, Aurousseau MH, Mal F, Meicler C, et al. Prospective study of screening for hepatocellular carcinoma in Caucasian patients with cirrhosis. J Hepatol 1994; 20: 65-71. [ Links ]
108. McMahon BJ, Alberts SR, Wainwright RB, Bulkow L, Lanier AP. Hepatitis B-related sequelae: prospective study in 1400 hepatitis B surface antigen-positive Alaska native carriers. Arch Intern Med 1990; 150: 1051-4. [ Links ]
109. McMahon BJ, Bulkow L, Harpster A, Snowball M, Lanier A, Sacco F, et al. Screening for hepatocellular carcinoma in Alaska natives infected with chronic hepatitis B: A 16-year populationbased study. Hepatology 2000; 32: 842-6. [ Links ]
110. Tanaka S, Kitamura T, Nakanishi K, Okuda S, Yamazaki H, Hiyama T, Fujimoto I. Effectiveness of periodic checkup by ultrasonography for the early diagnosis of hepatocellular carcinoma. Cancer 1990; 66: 22104. [ Links ]
111. Solmi L, Primerano AM, Gandolfi L. Ultrasound follow-up of patients at risk for hepatocellular carcinoma: results of a prospective study on 360 cases. Am J Gastroenterol 1996; 91: 1189-94. [ Links ]
112. Yuen MF, Cheng CC, Lauder IJ, Lam SK, Ooi CG, Lai CL. Early detection of hepatocellular carcinoma increases the chance of treatment: Hong Kong experience. Hepatology 2000; 31: 330-5. [ Links ]
113. Yuen MF, Lai CL. Screening for hepatocellular carcinoma: survival benefit and cost-effectiveness. Ann Oncol 2003; 14: 1463-7. [ Links ]
114. Wong LL, Limm WM, Severino R, Wong LM. Improved survival with screening for hepatocellular carcinoma. Liver Transpl 2000; 6: 320-5. [ Links ]
115. Sangiovanni A, Del NE, Fasani P, De FC, Ronchi G, Romeo R, et al. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology 2004; 126: 1005-14. [ Links ]
116. Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004; 130: 417-22. [ Links ]
117. Chen JG, Parkin DM, Chen QG, Lu JH, Shen QJ, Zhang BC, Zhu YR. Screening for liver cancer: results of a randomised controlled trial in Qidong, China. J Med Screen 2003; 10: 204-9. [ Links ]
118. Chalasani N, Said A, Ness R, Hoen H, Lumeng L. Screening for hepatocellular carcinoma in patients with cirrhosis in the United States: results of a national survey. Am J Gastroenterol 1999; 94: 2224-9. [ Links ]
119. Nguyen MH, Keeffe EB. Screening for hepatocellular carcinoma. J Clin Gastroenterol 2002; 35: S86-S91. [ Links ]
120. Lin OS, Keeffe EB, Sanders GD, Owens DK. Cost-effectiveness of screening for hepatocellular carcinoma in patients with cirrhosis due to chronic hepatitis C. Aliment Pharmacol Ther 2004; 19: 1159-72. [ Links ]
121. Thompson Coon J, Rogers G, Hewson P, Wright D, Anderson R, Cramp M, et al. Surveillance of cirrhosis for hepatocellular carcinoma: systematic review and economic analysis. Health Technol Assess 2007; 11: 1-206. [ Links ]
122. Thompson Coon J, Rogers G, Hewson P, Wright D, Anderson R, Jackson S, et al. Surveillance of cirrhosis for hepatocellular carcinoma: a cost-utility analysis. Br J Cancer 2008; 98: 1166-75. [ Links ]
123. Oka H, Tamori A, Kuroki T, Kobayashi K, Yamamoto S. Prospective study of alpha-fetoprotein in cirrhotic patients monitored for development of hepatocellular carcinoma. Hepatology 1994; 19: 61-6. [ Links ]
124. Colombo M. Screening for cancer in viral hepatitis. Clin Liver Dis 2001; 5: 109-22. [ Links ]
125. Bonis PA, Tong MJ, Blatt LM, Conrad A, Griffith JL. A predictive model for the development of hepatocellular carcinoma, liver failure, or liver transplantation for patients presenting to clinic with chronic hepatitis C. Am J Gastroenterol 1999; 94: 1605-12. [ Links ]
126. Velazquez RF, Rodriguez M, Navascues CA, Linares A, Perez R, Sotorrios NG, et al. Prospective analysis of risk factors for hepatocellular carcinoma in patients with liver cirrhosis. Hepatology 2003; 37: 520-7. [ Links ]
127. Kudo M, Okanoue T. Management of hepatocellular carcinoma in Japan: Consensus-based clinical practice manual proposed by the Japan Society of Hepatology. Oncology 2007; 72: 2-15 (suppl 1). [ Links ]
128. Makuuchi M, Kokudo N, Arii S, Futagawa S, Kaneko S, Kawasaki S, et al. Development of evidence-based clinical guidelines for the diagnosis and treatment of hepatocellular carcinoma in Japan. Hepatol Res 2008; 38: 37-51. [ Links ]
129. Arase Y, Ikeda K, Suzuki F, Suzuki Y, Kobayashi M, Akuta N, et al. Prolonged-interferon therapy reduces hepatocarcinogenesis in aged-patients with chronic hepatitis C. J Med Virol 2007; 79: 1095-102. [ Links ]
130. Murashima S, Tanaka M, Haramaki M, Yutani S, Nakashima Y, Harada K, et al. A decrease in AFP level related to administration of interferon in patients with chronic hepatitis C and a high level of AFP. Dig Dis Sci 2006; 51: 808-12. [ Links ]
131. Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol 2001; 35: 421-30. [ Links ]
132. Giovannini M, Elias D, Monges G, Raoul JL, Rougier P. Hepatocellular carcinoma. Br J Cancer 2001;84:74-77 (suppl 2). [ Links ]
133. Bialecki ES, Ezenekwe AM, Brunt EM, Collins BT, Ponder TB, Bieneman BK, Di Bisceglie AM. Comparison of liver biopsy and noninvasive methods for diagnosis of hepatocellular carcinoma. Clin Gastroenterol Hepatol 2006; 4: 361-8. [ Links ]
134. Sturgeon CM, Lai LC, Duffy MJ. Serum tumour markers: How to order and interpret them. BMJ 2009; 339: b3527. [ Links ]
135. National Comprehensive Cancer Network (NCCN) clinical practice guidelines in oncology, hepatobiliary cancers V.I. 2010. http://www.nccn.org/ [ Links ]
136. Poon D, Anderson BO, Chen LT, Tanaka K, Lau WY, Van Cutsem E, et al. Management of hepatocellular carcinoma in Asia: consensus statement from the Asian Oncology Summit 2009. Lancet Oncol 2009; 10: 1111-8. [ Links ]
137. European Group on Tumor Markers (EGTM): Consensus recommendations. Anticancer Res 1999; 19: 2785-820. [ Links ]
138. Sturgeon CM, Duffy MJ, Stenman UH, Lilja H, Brunner N, Chan DW, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in testicular, prostate, colorectal, breast, and ovarian cancers. Clin Chem 2008; 54: e11-79. [ Links ]
139. McIntire KR, Waldmann TA, Moertel CG, Go VL. Serum alphafetoprotein in patients with neoplasms of the gastrointestinal tract. Cancer Res 1975; 35: 991-6. [ Links ]
140. Sawabu N, Hattori N, Okuda K, Ishak KG. Serological tumor markers in hepatocellular carcinoma. In: Okuda K, Ishak KG, eds. Neoplasms of the liver. Tokyo, Japan, Springer-Verlag; 1987, pp. 227-38. [ Links ]
141. Wu JT. Serum alpha-fetoprotein and its lectin reactivity in liver diseases: A review. Ann Clin Lab Sci 1990; 20: 98-105. [ Links ]
142. Daniele B, Bencivenga A, Megna AS, Tinessa V. Alpha-fetoprotein and ultrasonography screening for hepatocellular carcinoma. Gastroenterology 2004; 127: S108- S112. [ Links ]
143. Talwalkar JA, Gores GJ. Diagnosis and staging of hepatocellular carcinoma. Gastroenterology 2004;127: S126-32. [ Links ]
144. Fujiyama S, Izuno K, Yamasaki K, Sato T, Taketa K. Determination of optimum cutoff levels of plasma desgamma- carboxy prothrombin and serum alpha-fetoprotein for the diagnosis of hepatocellular carcinoma using receiver operating characteristic curves. Tumour Biol 1992; 13: 316-23. [ Links ]
145. Liver cancer study group of Japan. The 15th report on Nationwide follow-up studies of primary liver cancer. Acta Hep Jap 2003; 44: 157-75. [ Links ]
146. Lee HS, Chung YH, Kim CY. Specificity of serum a-fetoprotein in HBsAg+ and HBxAg- patients in the diagnosis of hepatocellular carcinoma. Hepatology 1991; 14: 68- 72. [ Links ]
147. Namieno T, Kawata A, Sato N, Kondo Y, Uchino J. Agerelated, different clinicopathologic features of hepatocellular carcinoma patients. Ann Surg 1995; 221: 308- 14. [ Links ]
148. Taketa K. Alpha-fetoprotein. J Med Tech 1989; 33: 1380-84. [ Links ]
149. The Liver Study Group of Japan: Primary cancer in Japan. Sixth Report. Cancer 1987; 60: 1400-11. [ Links ]
150. Matsui H, Rimal N, Kamakura K, Uesugi S, Yamamoto H, Ikeda S, Taketa K. Serum alpha-fetoprotein levels in healthy Japanese adults. Acta Med Okayama 1998; 52: 149-54. [ Links ]
151. Greene FL, Page DL, Fleming ID, et al. AJCC (American Joint Committee on Cancer) Cancer Staging Manual. Vol. 6. New York, NY, Springer-Velag; 2002, pp 131-8. [ Links ]
152. Okuda K, Ohtsuki T, Obata H, Tomimatsu M, Okazaki N, Hasegawa H, et al. Natural history of hepatocellular carcinoma and prognosis in relation to treatment. Study of 850 patients. Cancer 1985; 56: 918-28. [ Links ]
153. Kudo M, Chung H, Haji S, Osaki Y, Oka H, Seki T, et al. Validation of a new prognostic staging system for hepatocellular carcinoma. The JIS score compared with the CLIP score. Hepatology 2004; 40: 1396-405. [ Links ]
154. Chevret S, Trinchet JC, Mathieu D, Rached AA, Beaugrand M, Chastang C. A new prognostic classification for predicting survival in patients with hepatocellular carcinoma. Groupe d'Etude et de Traitement du Carcinome Hepatocellulaire. J Hepatol 1999; 31: 133-41. [ Links ]
155. Prospective validation of the CLIP score: A new prognostic system for patients with cirrhosis and hepatocellular carcinoma. The Cancer of the Liver Italian Program (CLIP) Investigators. Hepatology 2000; 31: 840-5. [ Links ]
156. Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma. The BCLC staging classification. Semin Liver Dis 1999; 19: 329-38. [ Links ]
157. Bruix J, Llovet JM. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology 2002; 35: 519-24. [ Links ]
158. Leung TW, Tang AM, Zee B, Lau WY, Lai PB, Leung KL, et al. Construction of the Chinese University Prognostic Index for hepatocellular carcinoma and comparison with the TNM staging system, the Okuda staging system, and the Cancer of the Liver Italian Program staging system. A study based on 926 patients. Cancer 2002; 94: 1760-9. [ Links ]
159. Llovet JM, Beaugrand M. Hepatocellular carcinoma. Present status and future prospects. J Hepatol 2003;38:S136-S49 (suppl 1). [ Links ]
160. Henderson JM, Sherman M, Tavill A, Abecassis M, Chejfec G, Gramlich R. AHPBA/AJCC consensus conference on staging of hepatocellular carcinoma: consensus statement HPB (Oxford) 2003; 5: 243-50. [ Links ]
161. Marrero JA, Fontana RJ, Barrat A, Askari F, Conjeevaram HS, Su GL, Lok AS. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology 2005; 41: 707-16. [ Links ]
162. Cho CS, Gonen M, Shia J, Kattan MW, Klimstra DS, Jarnagin WR, et al. A novel prognostic nomogram is more accurate than convencional staging systems for predicting survival after resection of hepatocellular carcinoma. J Am Coll Surg 2008; 206: 281-91. [ Links ]
163. Lerose R, Molinari R, Rocchi E, Manenti F, Villa E. Prognostic features and survival of hepatocellular carcinoma in Italy: impact of stage of disease. Eur J Cancer 2001; 37: 239-45. [ Links ]
164. Ikai I, Arii S, Kojiro M, Ichida T, Makuuchi M, Matsuyama Y, et al. Reevaluation of prognostic factors for survival after liver resection in patients with hepatocellular carcinoma in a Japanese nationwide survey. Cancer 2004; 101: 796-802. [ Links ]
165. Shiraki K, Takase K, Tameda Y, Hamada M, Kosaka Y, Nakano T. A clinical study of lectin-reactive alpha-fetoprotein as an early indicator of hepatocellular carcinoma in the follow-up of cirrhotic patients. Hepatology 1995; 22: 802-7. [ Links ]
166. Tangkijvanich P, Anukulkarnkusol N, Suwangool P, Lertmaharit S, Hanvivatvong O, Kullavanijaya P, Poovorawan Y. Clinical characteristics and prognosis of hepatocellular carcinoma: analysis based on serum alpha-fetoprotein levels. J Clin Gastroenterol 2000; 31: 302-8. [ Links ]
167. Farinati F, Marino D, De Giorgio M, Baldan A, Cantarini M, Cursaro C, et al. Diagnostic and prognostic role of alpha- fetoprotein in hepatocellular carcinoma: both or neither? Am J Gastroenterol 2006; 101: 524-32. [ Links ]
168. Andorno E, Salizzoni M, Schieroni R, De HB. Role of serum alpha-fetoprotein in pre- and post-orthotopic liver transplantation (OLT) for malignant disease. J Nucl Med Allied Sci 1989; 33: 132-4. [ Links ]
169. Ebara M, Ohto M, Shinagawa T, Sugiura N, Kimura K, Matsutani S, et al. Natural history of minute hepatocellular carcinoma smaller than three centimeters complicating cirrhosis. A study in 22 patients. Gastroenterology 1986; 90: 289-98. [ Links ]
170. Matsumoto Y, Suzuki T, Asada I, Ozawa K, Tobe T, Honjo I. Clinical classification of hepatoma in Japan according to serial changes in serum alpha-fetoprotein levels. Cancer 1982; 49: 354-60. [ Links ]
171. Scottish Hepatopancreatobiliary Managed Clinical Network: Guidelines for the management of hepatocelluar carcinoma. www.scan.scot.nhs.uk [ Links ]
172. Johnson PJ, Williams R. Serum alpha-fetoprotein estimations and doubling time in hepatocellular carcinoma. Influence of therapy and possible value in early detection. J Natl Cancer Inst 1980; 64: 1329-32. [ Links ]
173. Toyoda H, Kumada T, Kaneoka Y, Osaki Y, Kimura T, Arimoto A, et al. Prognostic value of pretreatment levels of tumor markers for hepatocellular carcinoma on survival after curative treatment of patients with HCC. J Hepatol 2008; 49: 223-32. [ Links ]
174. Urabe T, Hayashi S, Terasaki S, Terada M, Matusushita E, Kaneko S, et al. An assessment of therapeutic effect of hepatocellular carcinoma by the serial changes in serum AFP value [Japanese]. Nippon Shokakibyo Gakkai Zasshi 1990; 87: 100-108. [ Links ]
175. McIntire KR, Vogel CL, Primack A, Waldmann TA, Kyalwazi SK. Effect of surgical and chemotherapeutic treatment on alphafetoprotein levels in patients with hepatocellular carcinoma. Cancer 1976; 37: 677-83. [ Links ]
176. Matsumoto Y, Suzuki T, Ono H, Nakase A, Honjo I. Response of alpha-fetoprotein to chemotherapy in patients with hepatomas. Cancer 1974;34:1602-1606. [ Links ]
177. Leung TW, Patt YZ, Lau WY, Ho SK, Yu SC, Chan AT, et al. Complete pathological remission is possible with systemic combination chemotherapy for inoperable hepatocellular carcinoma. Clin Cancer Res 1999; 5: 1676-81. [ Links ]
178. Nauta RJ, Heres EK, Thomas DS, Harter KW, Rodgers JE, Holt RW, et al. Intraoperative single-dose radiotherapy. Observations on staging and interstitial treatment of unresectable liver metastases. Arch Surg 1987; 122: 1392-5. [ Links ]
179. Chan SL, Mo FK, Johnson PJ, Hui EP, Ma BB, Ho WM, et al. New utility of an old marker: serial alpha-fetoprotein measurement in predicting radiologic response and survival of patients with hepatocellular carcinoma undergoing systemic chemotherapy. J Clin Oncol 2009; 27: 446-52. [ Links ]
180. Vora SR, Zheng H, Stadler ZK, Fuchs CS, Zhu AX. Serum alpha-fetoprotein response as a surrogate for clinical outcome in patients receiving systemic therapy for advanced hepatocellular carcinoma. Oncologist 2009; 14: 717-25. [ Links ]
181. Liebman HA, Furie BC, Tong MJ, Blanchard RA, Lo KJ, Lee SD, et al. Des-gamma-carboxy (abnormal) prothrombin as a serum marker of primary hepatocellular carcinoma. N Engl J Med 1984; 310: 1427-31. [ Links ]
182. Weitz IC, Liebman HA. Des-gamma-carboxy (abnormal) prothrombin and hepatocellular carcinoma. A critical review. Hepatology 1993; 18: 990-7. [ Links ]
183. Mita Y, Aoyagi Y, Yanagi M, Suda T, Suzuki Y, Asakura H. The usefulness of determining des-gamma-carboxy prothrombin by sensitive enzyme immunoassay in the early diagnosis of patients with hepatocellular carcinoma. Cancer 1998; 82: 1643-8. [ Links ]
184. Okuda H, Nakanishi T, Takatsu K, Saito A, Hayashi N, Watanabe K, et al. Measurement of serum levels of desgamma- carboxy prothrombin in patients with hepatocellular carcinoma by a revised enzyme immunoassay kit with increased sensitivity. Cancer 1999; 85: 812-8. [ Links ]
185. Tanaka Y, Kashiwagi T, Tsutsumi H, Nagasawa M, Toyama T, Ozaki S, et al. Sensitive measurement of serum abnormal prothrombin (PIVKA-II) as a marker of hepatocellular carcinoma. Hepatogastroenterology 1999; 46: 2464-8. [ Links ]
186. Okuda H, Nakanishi T, Takatsu K, Saito A, Hayashi N, Takasaki K, et al. Serum levels of des-gamma-carboxy prothrombin measured using the revised enzyme immunoassay kit with increased sensitivity in relation to clinicopathologic features of solitary hepatocellular carcinoma. Cancer 2000; 88: 544-9. [ Links ]
187. Marrero JA, Su GL, Wei W, Emick D, Conjeevaram HS, Fontana RJ, Lok AS. Des-gamma carboxyprothrombin can differentiate hepatocellular carcinoma from nonmalignant chronic liver disease in American patients. Hepatology 2003; 37: 1114-21. [ Links ]
188. Koike Y, Shiratori Y, Sato S, Obi S, Teratani T, Imamura M, et al. Des-gamma-carboxy prothrombin as a useful predisposing factor for the development of portal venous invasion in patients with hepatocellular carcinoma. A prospective analysis of 227 patients. Cancer 2001; 91: 561-9. [ Links ]
189. Imamura H, Matsuyama Y, Miyagawa Y, Ishida K, Shimada R, Miyagawa S, et al. Prognostic significance of anatomical resection and des-gamma-carboxy prothrombin in patients with hepatocellular carcinoma. Br J Surg 1999; 86: 1032-8. [ Links ]
190. Hamamura K, Shiratori Y, Shiina S, Imamura M, Obi S, Sato S, et al. Unique clinical characteristics of patients with hepatocellular carcinoma who present with high plasma des-gammacarboxy prothrombin and low serum alpha-fetoprotein. Cancer 2000; 88: 1557-64. [ Links ]
191. Nakamura S, Nouso K, Sakaguchi K, Ito YM, Ohashi Y, Kobayashi Y, et al. Sensitivity and specificity of desgamma- carboxy prothrombin for diagnosis of patients with hepatocellular carcinomas varies according to tumor size. Am J Gastroenterol 2006; 101: 2038-43. [ Links ]
192. Kobayashi M, Ikeda K, Kawamura Y, Yatsuji H, Hosaka T, Sezaki H, et al. High serum des-gamma-carboxy prothrombin level predicts poor prognosis after radiofrequency ablation of hepatocellular carcinoma. Cancer 2009; 115: 571- 80. [ Links ]
193. Durazo FA, Blatt LM, Corey WG, Lin JH, Han S, Saab S, et al. Des-gamma-carboxyprothrombin, alpha-fetoprotein and AFP-L3 in patients with chronic hepatitis, cirrosis and hepatocellular carcinoma. J Gastroenterol Hepatol 2008; 23: 1541-8. [ Links ]
194. Fujiyama S, Tanaka M, Maeda S, Ashihara H, Hirata R, Tomita K. Tumor markers in early diagnosis, follow-up and Management of patients with hepatocellular carcinoma. Oncology 2002; 62: 57-63 (suppl 1). [ Links ]
195. Hsu HC, Cheng W, Lai PL. Cloning and expression of a developmentally regulated transcript MXR7 in hepatocellular carcinoma: biological significance and temporospatial distribution. Cancer Res 1997; 57: 5179-84. [ Links ]
196. Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, Filmus J. Glypican-3. A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003; 125: 89-97. [ Links ]
197. Sung YK, Hwang SY, Park MK, Farooq M, Han IS, Bae HI, et al. Glypican-3 is overexpressed in human hepatocellular carcinoma. Cancer Sci 2003; 94: 259-62. [ Links ]
198. Capurro M, Filmus J. Glypican-3 as a serum marker for hepatocellular carcinoma. Cancer Res 2005; 65: 372; author reply 372-3. [ Links ]
199. Hippo Y, Watanabe K, Watanabe A, Midorikawa Y, Yamamoto S, Ihara S, et al. Identification of soluble NH2- terminal fragment of glypican-3 as a serological marker for early-stage hepatocellular carcinoma. Cancer Res 2004; 64: 2418-23. [ Links ]
200. Matsumura M, Shiratori Y, Niwa Y, Tanaka T, Ogura K, Okudaira T, et al. Presence of alpha-fetoprotein mRNA in blood correlatos with outcome in patients with hepatocellular carcinoma. J Hepatol 1999; 31: 332-9. [ Links ]
201. Ijichi M, Takayama T, Matsumura M, Shiratori Y, Omata M, Makuuchi M. alpha-fetoprotein mRNA in the circulation as a predictor of postsurgical recurrence of hepatocellular carcinoma. A prospective study. Hepatology 2002; 35: 853-60. [ Links ]
202. Lemoine A, Le BT, Salvucci M, Azoulay D, Pham P, Raccuia J, et al. Prospective evaluation of circulating hepatocytes by alphafetoprotein mRNA in humans during liver surgery. Ann Surg 1997; 226: 43-50. [ Links ]
203. Witzigmann H, Geissler F, Benedix F, Thiery J, Uhlmann D, Tannapfel A, et al. Prospective evaluation of circulating hepatocytes by alpha-fetoprotein messenger RNA in patients with hepatocellular carcinoma. Surgery 2002; 131: 34-43. [ Links ]
204. Iavarone M, Lampertico P, Ronchi G, Del NE, Zanella A, Colombo M. A prospective study of blood alpha-fetoprotein messenger RNA as a predictor of hepatocellular carcinoma in patients with cirrhosis. J Viral Hepat 2003; 10: 423-6. [ Links ]
205. Katoh H, Ojima H, Kokubu A, Saito S, Kondo T, Kosuge T, et al. Genetically distinct and clinically relevant classification of hepatocellular carcinoma: putative therapeutic targets. Gastroenterology 2007; 133: 1475-86. [ Links ]
206. Kittaka N, Takemasa I, Takeda Y, Marubashi S, Nagano H, Umeshita K, et al. Molecular mapping of human hepatocellular carcinoma provides deeper biological insight from genomic data. Eur J Cancer 2008; 44: 885- 97. [ Links ]
207. Mann CD, Neal CP, Garcea G, Manson MM, Dennison AR, Berry DP. Prognostic molecular markers in hepatocellular carcinoma: a systematic review. Eur J Cancer 2007; 43: 979-92. [ Links ]
208. Sun S, Lee NP, Poon RT, Fan ST, He QY, Lau GK, Luk JM. Oncoproteomics of hepatocellular carcinoma: from cancer markers' discovery to functional pathways. Liver Int 2007; 27: 1021-38. [ Links ]
209. Zinkin NT, Grall F, Bhaskar K, Otu HH, Spentzos D, Kalmowitz B, et al. Serum proteomics and biomarkers in hepatocellular carcinoma and chronic liver disease. Clin Cancer Res 2008; 14: 470-7. [ Links ]
210. Lo YM. Circulating nucleic acids in plasma and serum: an overview. Ann N Y Acad Sci 2001; 945: 1-7. [ Links ]
211. Wong IH, Lo YM, Zhang J, Liew CT, Ng MH, Wong N, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res 1999; 59: 71-3. [ Links ]
212. Lee HS, Kim BH, Cho NY, Yoo EJ, Choi M, Shin SH, et al. Prognostic implications of and relationship between CpG island hypermethylation and repetitive DNA hypomethylation in hepatocellular carcinoma. Clin Cancer Res 2009; 15: 812-20. [ Links ]
213. Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 2008; 47: 1955-63. [ Links ]
214. Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, et al. Liver stem cells and hepatocellular carcinoma. Hepatology 2009; 49: 318-29. [ Links ]
215. Smith MW, Yue ZN, Geiss GK, Sadovnikova NY, Carter VS, Boix L, et al. Identification of novel tumor markers in hepatitis C virus-associated hepatocellular carcinoma. Cancer Res 2003; 63: 859-64. [ Links ]
216. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin 2009; 59: 225-49. [ Links ]
217. Vineis P, Esteve J, Hartge P, Hoover R, Silverman DT, Terracini B. Effects of timing and type of tobacco in cigarette-induced bladder cancer. Cancer Res 1988; 48: 3849-52. [ Links ]
218. Lamm DL, Torti FM. Bladder cancer, 1996. CA Cancer J Clin 1996; 46: 93-112. [ Links ]
219. Bryan RT, Wallace DM. 'Superficial' bladder cancer-time to uncouple pT1 tumours from pTa tumours. BJU Int 2002; 90: 846-52. [ Links ]
220. Sauter G, Algaba F, Amin M. Tumours of the urinary system: noninvasive urothelial neoplasias. Lyon, France, IARCC Press, 2004, pp 29-34. [ Links ]
221. Busch C, Algaba F. The WHO/ISUP 1998 and WHO 1999 systems for malignancy grading of bladder cancer. Scientific foundation and translation to one another and previous systems. Virchows Arch 2002; 441:105-8. [ Links ]
222. Agarwal PK, Black PC, Kamat AM. Considerations on the use of diagnostic markers in management of patients with bladder cancer. World J Urol 2008; 26: 39-44. [ Links ]
223. Theodorescu D, Wittke S, Ross MM, Walden M, Conaway M, Just I, et al. Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol 2006; 7: 230-40. [ Links ]
224. Sanchez-Carbayo M, Cordon-Cardo C. Molecular alterations associated with bladder cancer progression. Semin Oncol 2007; 34: 75-84. [ Links ]
225. Ecke TH. Focus on urinary bladder cancer markers: a review. Minerva Urol Nefrol 2008; 60: 237-46. [ Links ]
226. Cordon-Cardo C, Cote RJ, Sauter G. Genetic and molecular markers of urothelial premalignancy and malignancy. Scand J Urol Nephrol 2000; 82-93 (suppl). [ Links ]
227. Wolff EM, Liang G, Jones PA. Mechanisms of disease. Genetic and epigenetic alterations that drive bladder cancer. Nat Clin Pract Urol 2005;2:502-510. [ Links ]
228. Vrooman OP, Witjes JA. Urinary markers in bladder cancer. Eur Urol 2008; 53: 909-16. [ Links ]
229. Droller MJ. Bladder cancer. State-of-the-art care. CA Cancer J Clin 1998; 48: 269-84. [ Links ]
230. Parmar MK, Freedman LS, Hargreave TB, Tolley DA. Prognostic factors for recurrence and followup policies in the treatment of superficial bladder cancer. Report from the British Medical Research Council Subgroup on Superficial Bladder Cancer (Urological Cancer Working Party). J Urol 1989; 142: 284-8. [ Links ]
231. Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al. Grading quality of evidence and strength of recommendations. BMJ 2004; 328: 1490. [ Links ]
232. National Comprehensive Cancer Network (NCCN). Clinical Practice Guidelines in Oncology: bladder cancer. V. 1.2010. http://www.nccn.org/professionals/physician_gls/PDF/bladder.pdf [ Links ]
233. Zipfel PF, Skerka C. Complement factor H and related proteins. An expanding family of complement-regulatory proteins? Immunol Today 1994; 15: 121-6. [ Links ]
234. Gutierrez Banos JL, Martin Garcia B, Hernandez Rodriguez R, Portillo Martin JA, Correas Gomez MA, del Valle Schaan JI, et al. Usefulness of BTA Stat test (Bard) in the diagnosis of bladder cancer. Preliminary results and comparison with cytology and cystoscopy. Arch Esp Urol 1998; 51: 778-82. [ Links ]
235. Sharma S, Zippe CD, Pandrangi L, Nelson D, Agarwal A. Exclusion criteria enhance the specificity and positive predictive value of NMP22 and BTA stat. J Urol 1999; 162: 53-7. [ Links ]
236. Takashi M, Schenck U, Kissel K, Leyh H, Treiber U. Use of diagnostic categories in urinary cytology in comparison with the bladder tumour antigen (BTA) test in bladder cancer patients. Int Urol Nephrol 1999; 31: 189-96. [ Links ]
237. Landman J, Chang Y, Kavaler E, Droller MJ, Liu BC. Sensitivity and specificity of NMP-22, telomerase, and BTA in the detection of human bladder cancer. Urology 1998; 52: 398-402. [ Links ]
238.Wiener HG, Mian C, Haitel A, Pycha A, Schatzl G, Marberger M. Can urine bound diagnostic tests replace cystoscopy in the management of bladder cancer? J Urol 1998; 159: 1876-80. [ Links ]
239. Leyh H, Marberger M, Conort P, Sternberg C, Pansadoro V, Pagano F, et al. Comparison of the BTA stat test with voided urine cytology and bladder wash cytology in the diagnosis and monitoring of bladder cancer. Eur Urol 1999; 35: 52-6. [ Links ]
240. Leyh H, Mazeman E. Bard BTA test compared with voided urine cytology in the diagnosis of recurrent bladder cancer. Eur Urol 1997; 32: 425-8. [ Links ]
241. Sarosdy MF, Hudson MA, Ellis WJ, Soloway MS, DeVere White R, Sheinfeld J, et al. Improved detection of recurrent bladder cancer using the Bard BTA stat Test. Urology 1997; 50: 349-53. [ Links ]
242. Thomas L, Leyh H, Marberger M, Bombardieri E, Bassi P, Pagano F, et al. Multicenter trial of the quantitative BTA TRAK assay in the detection of bladder cancer. Clin Chem 1999; 45: 472-7. [ Links ]
243. Mattioli S, Seregni E, Caperna L, Botti C, Savelli G, Bombardieri E. BTA-TRAK combined with urinary cytology is a reliable urinary indicator of recurrent transitional cell carcinoma (TCC) of the bladder. Int J Biol Markers 2000; 15: 219-25. [ Links ]
244. Herman MP, Svatek RS, Lotan Y, Karakiewizc PI, Shariat SF. Urine-based biomarkers for the early detection and surveillance of non-muscle invasive bladder cancer. Minerva Urol Nefrol 2008; 60: 217-35. [ Links ]
245. Soloway MS, Briggman V, Carpinito GA, Chodak GW, Church PA, Lamm DL, et al. Use of a new tumor marker, urinary NMP22, in the detection of occult or rapidly recurring transitional cell carcinoma of the urinary tract following surgical treatment. J Urol 1996; 156: 363-7. [ Links ]
246. Miyanaga N, Akaza H, Ishikawa S, Ohtani M, Noguchi R, Kawai K, et al. Clinical evaluation of nuclear matrix protein 22 (NMP22) in urine as a novel marker for urothelial cancer. Eur Urol 1997; 31: 163-8. [ Links ]
247.Miyanaga N, Akaza H, Tsukamoto S, Shimazui T, Ohtani M, Ishikawa S, et al. Usefulness of urinary NMP22 to detect tumor recurrence of superficial bladder cancer after transurethral resection. Int J Clin Oncol 2003; 8: 369-73. [ Links ]
248. Stampfer DS, Carpinito GA, Rodriguez-Villanueva J, Willsey LW, Dinney CP, Grossman HB, et al. Evaluation of NMP22 in the detection of transitional cell carcinoma of the bladder. J Urol 1998; 159: 394-8. [ Links ]
249. Lahme S, Bichler KH, Feil G, Zumbragel A, Gotz T. Comparison of cytology and nuclear matrix protein 22 (NMP 22) for the detection and follow-up of bladder-cancer. Adv Exp Med Biol 2003; 539: 111-9. [ Links ]
250. Ponsky LE, Sharma S, Pandrangi L, Kedia S, Nelson D, Agarwal A, Zippe CD. Screening and monitoring for bladder cancer: refining the use of NMP22. J Urol 2001; 166: 75-8. [ Links ]
251. Tomera KM. NMP22 BladderChek Test: point-of-care technology with life- and money-saving potential. Expert Rev Mol Diagn 2004; 4: 783-94. [ Links ]
252. Yokoyama T, Sekigawa R, Hayashi T, Horita S, Kanamuro T, Nonami Y, et al. The clinical efficacy of Bladder Chek NMP22 in urothelial cancer. Rinsho Byori 2004; 52: 199-203. [ Links ]
253. Atsu N, Ekici S, Oge OO, Ergen A, Hascelik G, Ozen H. Falsepositive results of the NMP22 test due to hematuria. J Urol 2002; 167: 555-8. [ Links ]
254. Grossman HB, Messing E, Soloway M, Tomera K, Katz G, Berger Y, et al. Detection of bladder cancer using a point-of-care proteomic assay. JAMA 2005; 293: 810-6. [ Links ]
255. Lokeshwar VB, Habuchi T, Grossman HB, Murphy WM, Hautmann SH, Hemstreet GP 3rd, et al. Bladder tumor markers beyond cytology: International Consensus Panel on bladder tumor markers. Urology 2005; 66: 35-63. [ Links ]
256. Grossman HB, Soloway M, Messing E, Katz G, Stein B, Kassabian V, Shen Y. Surveillance for recurrent bladder cancer using a point-of-care proteomic assay. JAMA 2006; 295: 299-305. [ Links ]
257. Mian C, Pycha A, Wiener H, Haitel A, Lodde M, Marberger M. Immunocyt: a new tool for detecting transitional cell cancer of the urinary tract. J Urol 1999; 161: 1486-9. [ Links ]
258. Messing EM, Teot L, Korman H, Underhill E, Barker E, Stork B, et al. Performance of urine test in patients monitored for recurrence of bladder cancer: a multicenter study in the United States. J Urol 2005; 174: 1238-41. [ Links ]
259. Toma MI, Friedrich MG, Hautmann SH, Jakel KT, Erbersdobler A, Hellstern A, et al. Comparison of the ImmunoCyt test and urinary cytology with other urine tests in the detection and surveillance of bladder cancer. World J Urol 2004; 22: 145-9. [ Links ]
260. Feil G, Zumbragel A, Paulgen-Nelde HJ, Hennenlotter J, Maurer S, Krause S, et al. Accuracy of the ImmunoCyt assay in the diagnosis of transitional cell carcinoma of the urinary bladder. Anticancer Res 2003; 23: 963-7. [ Links ]
261. Lodde M, Mian C, Negri G, Berner L, Maffei N, Lusuardi L, et al. Role of uCyt+ in the detection and surveillance of urothelial carcinoma. Urology 2003; 61: 243-7. [ Links ]
262. Halling KC, King W, Sokolova IA, Meyer RG, Burkhardt HM, Halling AC, et al. A comparison of cytology and fluorescence in situ hybridization for the detection of urothelial carcinoma. J Urol 2000; 164: 1768-75. [ Links ]
263. Sarosdy MF, Schellhammer P, Bokinsky G, Kahn P, Chao R, Yore L, et al. Clinical evaluation of a multi-target fluorescent in situ hybridization assay for detection of bladder cancer. J Urol 2002; 168: 1950-4. [ Links ]
264. Friedrich MG, Toma MI, Hellstern A, Pantel K, Weisenberger DJ, Noldus J, Huland H. Comparison of multitarget fluorescence in situ hybridization in urine with other noninvasive tests for detecting bladder cancer. BJU Int 2003; 92: 911-4. [ Links ]
265. Bollmann D, Bollmann M, Bankfalvi A, Heller H, Bollmann R, Pajor G, Hildenbrand R. Quantitative molecular grading of bladder tumours: a tool for objective assessment of the biological potential of urothelial neoplasias. Oncol Rep 2009; 21: 39-47. [ Links ]
266. Kipp BR, Karnes RJ, Brankley SM, Harwood AR, Pankratz VS, Sebo TJ, et al. Monitoring intravesical therapy for superficial bladder cancer using fluorescence in situ hybridization. J Urol 2005; 173: 401-4. [ Links ]
267. Gudjonsson S, Isfoss BL, Hansson K, Domanski AM, Warenholt J, Soller W, et al. The value of the UroVysion assay for surveillance of non-muscle-invasive bladder cancer. Eur Urol 2008; 54: 402-8. [ Links ]
268. Skacel M, Fahmy M, Brainard JA, Pettay JD, Biscotti CV, Liou LS, et al. Multitarget fluorescence in situ hybridization assay detects transitional cell carcinoma in the majority of patients with bladder cancer and atypical or negative urine cytology. J Urol 2003; 169: 2101-5. [ Links ]
269. Pycha A, Lodde M, Comploj E, Negri G, Egarter-Vigl E, Vittadello F, et al. Intermediate-risk urothelial carcinoma: an unresolved problem? Urology 2004; 63: 472-5. [ Links ]
270.Mengual L, Marin-Aguilera M, Ribal MJ, Burset M, Villavicencio H, Oliver A, et al. Clinical utility of fluorescence in situ hybridization for the surveillance of bladder cancer patients treated with bacillus Calmette-Guerin therapy. Eur Urol 2007; 52: 752-9. [ Links ]
271. Whitson J, Berry A, Carroll P, Konety B. A multicolour fluorescence in situ hybridization test predicts recurrence in patients with high-risk superficial bladder tumours undergoing intravesical therapy. BJU Int 2009; 104: 336-9. [ Links ]
272. Southgate J, Harnden P, Trejdosiewicz LK. Cytokeratin expression patterns in normal and malignant urothelium: a review of the biological and diagnostic implications. Histol Histopathol 1999; 14: 657-64. [ Links ]
273. Nisman B, Barak V, Shapiro A, Golijanin D, Peretz T, Pode D. Evaluation of urine CYFRA 21-1 for the detection of primary and recurrent bladder carcinoma. Cancer 2002; 94: 2914-22. [ Links ]
274. Pariente JL, Bordenave L, Jacob F, Gobinet A, Leger F, Ferriere JM, Le Guillou M. Analytical and prospective evaluation of urinary cytokeratin 19 fragment in bladder cancer. J Urol 2000; 163: 1116-9. [ Links ]
275. Siracusano S, Niccolini B, Knez R, Tiberio A, Benedetti E, Bonin S, et al. The simultaneous use of telomerase, cytokeratin 20 and CD4 for bladder cancer detection in urine. Eur Urol 2005; 47: 327-33. [ Links ]
276. Sanchez-Carbayo M, Herrero E, Megias J, Mira A, Soria F. Comparative sensitivity of urinary CYFRA 21-1, urinary bladder cancer antigen, tissue polypeptide antigen, tissue polypeptide antigen and NMP22 to detect bladder cancer. J Urol 1999; 162: 1951-6. [ Links ]
277. Sanchez-Carbayo M, Urrutia M, Silva JM, Romani R, Garcia J, Alferez F, et al. Urinary tissue polypeptide-specific antigen for the diagnosis of bladder cancer. Urology 2000; 55: 526-32. [ Links ]
278. Mian C, Lodde M, Haitel A, Vigl EE, Marberger M, Pycha A. Comparison of the monoclonal UBC-ELISA test and the NMP22 ELISA test for the detection of urothelial cell carcinoma of the bladder. Urology 2000; 55: 223-6. [ Links ]
279. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994; 266: 2011-5. [ Links ]
280. Yoshida K, Sugino T, Tahara H, Woodman A, Bolodeoku J, Nargund V, et al. Telomerase activity in bladder carcinoma and its implication for noninvasive diagnosis by detection of exfoliated cancer cells in urine. Cancer 1997; 79: 362-9. [ Links ]
281. Muller M, Krause H, Heicappell R, Tischendorf J, Shay JW, Miller K. Comparison of human telomerase RNA and telomerase activity in urine for diagnosis of bladder cancer. Clin Cancer Res 1998; 4: 1949-54. [ Links ]
282. de Kok JB, Ruers TJ, van Muijen GN, van Bokhoven A, Willems HL, Swinkels DW. Real-time quantification of human telomerase reverse transcriptase mRNA in tumors and healthy tissues. Clin Chem 2000; 46: 313-8. [ Links ]
283. Sanchini MA, Bravaccini S, Medri L, Gunelli R, Nanni O, Monti F, et al. Urine telomerase: an important marker in the diagnosis of bladder cancer. Neoplasia 2004; 6: 234-9. [ Links ]
284. Saad A, Hanbury DC, McNicholas TA, Boustead GB, Morgan S, Woodman AC. A study comparing various noninvasive methods of detecting bladder cancer in urine. BJU Int 2002; 89: 369-73. [ Links ]
285. Lee MY, Tsou MH, Cheng MH, Chang DS, Yang AL, Ko JS. Clinical application of NMP22 and urinary cytology in patients with hematuria or a history of urothelial carcinoma. World J Urol 2000; 18: 401-5. [ Links ]
286. Konety BR, Nguyen TS, Dhir R, Day RS, Becich MJ, Stadler WM, Getzenberg RH. Detection of bladder cancer using a novel nuclear matrix protein, BLCA-4. Clin Cancer Res 2000; 6: 2618-25. [ Links ]
287. Van Le TS, Myers J, Konety BR, Barder T, Getzenberg RH. Functional characterization of the bladder cancer marker, BLCA-4. Clin Cancer Res 2004; 10: 1384-91. [ Links ]
288. Konety BR, Nguyen TS, Brenes G, Sholder A, Lewis N, Bastacky S, et al. Clinical usefulness of the novel marker BLCA-4 for the detection of bladder cancer. J Urol 2000; 164: 634-9. [ Links ]
289. Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3: 917-21. [ Links ]
290. Dabrowski A, Filip A, Zgodzinski W, Dabrowska M, Polanska D, Wojcik M, et al. Assessment of prognostic significance of cytoplasmic survivin expression in advanced oesophageal cancer. Folia Histochem Cytobiol 2004; 42: 169-72. [ Links ]
291. Smith SD, Wheeler MA, Plescia J, Colberg JW, Weiss RM, Altieri DC. Urine detection of survivin and diagnosis of bladder cancer. Jama 2001; 285: 324-8. [ Links ]
292.Moussa O, Abol-Enein H, Bissada NK, Keane T, Ghoneim MA, Watson DK. Evaluation of survivin reverse transcriptasepolymerase chain reaction for noninvasive detection of bladder cancer. J Urol 2006; 175: 2312-6. [ Links ]
293. Lehner R, Lucia MS, Jarboe EA, Orlicky D, Shroyer AL, McGregor JA, et al. Immunohistochemical localization of the IAP protein survivin in bladder mucosa and transitional cell carcinoma. Appl Immunohistochem Mol Morphol 2002; 10: 134-8. [ Links ]
294. Pina-Cabral L, Santos L, Mesquita B, Amaro T, Magalhaes S, Criado B. Detection of survivin mRNA in urine of patients with superficial urothelial cell carcinomas. Clin Transl Oncol 2007; 9: 731-6. [ Links ]
295. Kenney DM, Geschwindt RD, Kary MR, Linic JM, Sardesai NY, Li ZQ. Detection of newly diagnosed bladder cancer, bladder cancer recurrence and bladder cancer in patients with hematuria using quantitative rt- PCR of urinary survivin. Tumour Biol 2007; 28: 57-62. [ Links ]
296. Shariat SF, Casella R, Khoddami SM, Hernandez G, Sulser T, Gasser TC, et al. Urine detection of survivin is a sensitive marker for the noninvasive diagnosis of bladder cancer. J Urol 2004; 171: 626-30. [ Links ]
297. Schultz IJ, Kiemeney LA, Karthaus HF, Witjes JA, Willems JL, Swinkels DW, et al. Survivin mRNA copy number in bladder washings predicts tumor recurrence in patients with superficial urothelial cell carcinomas. Clin Chem 2004; 50: 1425-8. [ Links ]
298. Schultz IJ, Wester K, Straatman H, Kiemeney LA, Babjuk M, Mares J, et al. Gene expression analysis for the prediction of recurrence in patients with primary Ta urothelial cell carcinoma. Eur Urol 2007; 51: 416- 22;discussion 422-3. [ Links ]
299. Simoneau M, Aboulkassim TO, LaRue H, Rousseau F, Fradet Y. Four tumor suppressor loci on chromosome 9q in bladder cancer: evidence for two novel candidate regions at 9q22.3 and 9q31. Oncogene 1999; 18: 157-63. [ Links ]
300. Czerniak B, Chaturvedi V, Li L, Hodges S, Johnston D, Roy JY, et al. Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer. Oncogene1999; 18: 1185-96. [ Links ]
301. Mao L, Schoenberg MP, Scicchitano M, Erozan YS, Merlo A, Schwab D, Sidransky D. Molecular detection of primary bladder cancer by microsatellite analysis. Science 1996; 271: 659-62. [ Links ]
302. Steiner G, Schoenberg MP, Linn JF, Mao L, Sidransky D. Detection of bladder cancer recurrence by microsatellite analysis of urine. Nat Med 1997; 3: 621-4. [ Links ]
303. von Knobloch R, Brandt H, Hofmann R. Molecular serological diagnosis in transitional cell bladder cancer. Ann N Y Acad Sci 2004; 1022: 70-5. [ Links ]
304. Fornari D, Steven K, Hansen AB, Vibits H, Jepsen JV, Poulsen AL, et al. Microsatellite analysis of urine sediment versus urine 44 Use of Tumor Markers in Liver, Bladder, Cervical, and Gastric Cancers cytology for diagnosing transitional cell tumors of the urinary bladder. APMIS 2004; 112: 148-52. [ Links ]
305. Utting M, Werner W, Dahse R, Schubert J, Junker K. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res 2002; 8: 35-40. [ Links ]
306. van der Aa MN, Zwarthoff EC, Steyerberg EW, Boogaard MW, Nijsen Y, van der Keur KA, et al. Microsatellite analysis of voided-urine samples for surveillence of low-grade non-muscleinvasive urothelial carcinoma. Feasibility and clinical utility in a prospective multicenter study (Cost-Effectiveness of Follow-Up of Urinary Bladder Cancer Trial [lsqb]CEFUB[rsqb]). Eur Urol 2008; 55: 659-67. [ Links ]
307. Lokeshwar VB, Obek C, Pham HT, Wei D, Young MJ, Duncan RC, et al. Urinary hyaluronic acid and hyaluronidase: markers for bladder cancer detection and evaluation of grade. J Urol 2000; 163: 348-56. [ Links ]
308. Pham HT, Block NL, Lokeshwar VB. Tumor-derived hyaluronidase: a diagnostic urine marker for high-grade bladder cancer. Cancer Res 1997; 57: 778-83. [ Links ]
309. Hautmann S, Toma M, Lorenzo Gomez MF, Friedrich MG, Jaekel T, Michl U, et al. Immunocyt and the HA-HAase urine tests for the detection of bladder cancer: a side-by-side comparison. Eur Urol 2004; 46: 466-71. [ Links ]
310. Schroeder GL, Lorenzo-Gomez MF, Hautmann SH, Friedrich MG, Ekici S, Huland H, et al. A side by side comparison of cytology and biomarkers for bladder cancer detection. J Urol 2004; 172: 1123-6. [ Links ]
311. Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet 1999;23:18-20. [ Links ]
312. Billerey C, Chopin D, Aubriot-Lorton MH, Ricol D, Gil Diez de Medina S, Van Rhijn B, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol 2001; 158: 1955-9. [ Links ]
313. Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000; 7: 165-97. [ Links ]
314. Horton WA, Lunstrum GP. Fibroblast growth factor receptor 3 mutations in achondroplasia and related forms of dwarfism. Rev Endocr Metab Disord 2002; 3: 381-5. [ Links ]
315. van Rhijn BW, Vis AN, van der Kwast TH, Kirkels WJ, Radvanyi F, Ooms EC, et al. Molecular grading of urothelial cell carcinoma with fibroblast growth factor receptor 3 and MIB-1 is superior to pathologic grade for the prediction of clinical outcome. J Clin Oncol 2003; 21: 1912-21. [ Links ]
316. van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC. The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res 2001; 61: 1265-8. [ Links ]
317. Habuchi T, Marberger M, Droller MJ, Hemstreet GP 3rd, Grossman HB, Schalken JA, et al. Prognostic markers for bladder cancer. International Consensus Panel on bladder tumor markers. Urology 2005; 66: 64-74. [ Links ]
318. Hernandez S, Lopez-Knowles E, Lloreta J, Kogevinas M, Amoros A, Tardon A, et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol 2006; 24: 3664-71. [ Links ]
319. Sawczuk IS, Pickens CL, Vasa UR, Ralph DA, Norris KA, Miller MC, et al. DD23 Biomarker. A prospective clinical assessment in routine urinary cytology specimens from patients being monitored for TCC. Urol Oncol 2002; 7: 185-90. [ Links ]
320. Gilbert SM, Veltri RW, Sawczuk A, Shabsigh A, Knowles DR, Bright S, et al. Evaluation of DD23 as a marker for detection of recurrent transitional cell carcinoma of the bladder in patients with a history of bladder cancer. Urology 2003; 61: 539-43. [ Links ]
321. Sanchez-Carbayo M, Urrutia M, Gonzalez de Buitrago JM, Navajo JA. Evaluation of two new urinary tumor markers. Bladder tumor fibronectin and cytokeratin 18 for the diagnosis of bladder cancer. Clin Cancer Res 2000; 6: 3585-94. [ Links ]
322. Hegele A, Heidenreich A, Varga Z, von Knobloch R, Olbert P, Kropf J, Hofmann R. Cellular fibronectin in patients with transitional cell carcinoma of the bladder. Urol Res 2003; 30: 363-6. [ Links ]
323. Hotakainen K, Haglund C, Paju A, Nordling S, Alfthan H, Rintala E, et al. Chorionic gonadotropin beta-subunit and core fragment in bladder cancer: mRNA and protein expresión in urine, serum and tissue. Eur Urol 2002; 41: 677-85. [ Links ]
324. Chan MW, Chan LW, Tang NL, Tong JH, Lo KW, Lee TL, et al. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin Cancer Res 2002; 8: 464-70. [ Links ]
325. Friedrich MG, Weisenberger DJ, Cheng JC, Chandrasoma S, Siegmund KD, Gonzalgo ML, et al. Detection of methylated apoptosis-associated genes in urine sediments of bladder cancer patients. Clin Cancer Res 2004; 10: 7457-65. [ Links ]
326. Dulaimi E, Uzzo RG, Greenberg RE, Al-Saleem T, Cairns P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin Cancer Res 2004; 10: 1887-93. [ Links ]
327. Vlahou A, Giannopoulos A, Gregory BW, Manousakas T, Kondylis FI, Wilson LL, et al. Protein profiling in urine for the diagnosis of bladder cancer. Clin Chem 2004; 50: 1438-41. [ Links ]
328. Zhang YF, Wu DL, Guan M, Liu WW, Wu Z, Chen YM, et al. Tree analysis of mass spectral urine profiles discriminates transitional cell carcinoma of the bladder from noncancer patient. Clin Biochem 2004; 37: 772-9. [ Links ]
329. Varkarakis MJ, Gaeta J, Moore RH, Murphy GP. Superficial bladder tumor. Aspects of clinical progression. Urology 1974; 4: 414-20. [ Links ]
330. Grossfeld GD, Litwin MS, Wolf JS, Hricak H, Shuler CL, Agerter DC, Carroll PR. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy-part I: definition, detection, prevalence, and etiology. Urology 2001; 57: 599-603. [ Links ]
331. Khadra MH, Pickard RS, Charlton M, Powell PH, Neal DE. A prospective analysis of 1,930 patients with hematuria to evaluate current diagnostic practice. J Urol 2000; 163: 524-7. [ Links ]
332. Mariani J. A prospective analysis of 1930 patients with hematuria to evaluate current diagnostic practice. J Urol 2000; 165: 545-6. [ Links ]
333. Grossfeld GD, Litwin MS, Wolf JS Jr, Hricak H, Shuler CL, Agerter DC, et al. Evaluation of asymptomatic microscopic hematuria in adults: the American Urological Association best practice policy-part II: patient evaluation, cytology, voided markers, imaging, cystoscopy, nephrology evaluation, and follow-up. Urology 2001; 57: 604-10. [ Links ]
334. Zippe C, Pandrangi L, Agarwal A. NMP22 is a sensitive, cost-effective test in patients at risk for bladder cancer. J Urol 1999; 161: 62-5. [ Links ]
335. Matritech I. PMA #940035. Submitted to the Food and Drug Administration;2000. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMA/pma.cfm?id=17679 [ Links ]
336. Stein JP, Grossfeld GD, Ginsberg DA, Esrig D, Freeman JA, Figueroa AJ, et al. Prognostic markers in bladder cancer: a contemporary review of the literature. J Urol 1998; 160: 645-59. [ Links ]
337. Zlotta AR, Schulman CC. Biological markers in superficial bladder tumors and their prognostic significance. Urol Clin North Am 2000; 27: 179-89, xi-xii. [ Links ]
338. Grossman HB, Liebert M, Antelo M, Dinney CP, Hu SX, Palmer JL, Benedict WF. p53 and RB expression predict progression in T1 bladder cancer. Clin Cancer Res 1998; 4: 829-34. [ Links ]
339. Lazar V, Diez SG, Laurent A, Giovangrandi Y, Radvanyi F, Chopin D, et al. Expression of human chorionic gonadotropina beta subunit genes in superficial and invasive bladder carcinomas. Cancer Res 1995; 55: 3735-8. [ Links ]
340. Gontero P, Banisadr S, Frea B, Brausi M. Metastasis markers in bladder cancer: a review of the literature and clinical considerations. Eur Urol 2004; 46: 296-311. [ Links ]
341. Esrig D, Spruck CH 3rd, Nichols PW, Chaiwun B, Steven K, Groshen S, et al. p53 nuclear protein accumulation correlatos with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am J Pathol 1993; 143: 1389-97. [ Links ]
342. Malats N, Bustos A, Nascimento CM, Fernandez F, Rivas M, Puente D, et al. P53 as a prognostic marker for bladder cancer: a meta-analysis and review. Lancet Oncol 2005; 6:678-86. [ Links ]
343. Gontero P, Casetta G, Zitella A, Ballario R, Pacchioni D, Magnani C, et al. Evaluation of P53 protein overexpression, Ki67 proliferative activity and mitotic index as markers of tumour recurrence in superficial transitional cell carcinoma of the bladder. Eur Urol 2000; 38: 287-96. [ Links ]
344. Vatne V, Maartmann-Moe H, Hoestmark J. The prognostic value of p53 in superficially infiltrating transitional cell carcinoma. Scand J Urol Nephrol 1995; 29: 491-5. [ Links ]
345. Cordon-Cardo C. Molecular alterations associated with bladder cancer initiation and progression. Scand J Urol Nephrol Suppl 2008: 154-65. [ Links ]
346. Ecke TH, Sachs MD, Lenk SV, Loening SA, Schlechte HH. TP53 gene mutations as an independent marker for urinary bladder cancer progression. Int J Mol Med 2008; 21: 655-61. [ Links ]
347. Sachs MD, Schlechte H, Lenk VS, Brenner S, Schnorr D, Fleige B, et al. Genetic analysis of Tp53 from urine sediment as a tool for diagnosing recurrence and residual of bladder carcinoma. Eur Urol 2000; 38: 426-33. [ Links ]
348. Schlichtholz B, Presler M, Matuszewski M. Clinical implications of p53 mutation analysis in bladder cancer tissue and urine sediment by functional assay in yeast. Carcinogenesis 2004; 25: 2319-23. [ Links ]
349. Aleman A, Cebrian V, Alvarez M, Lopez V, Orenes E, Lopez- Serra L, et al. Identification of PMF1 methylation in association with bladder cancer progression. Clin Cancer Res 2008; 14: 8236-43. [ Links ]
350. Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon- Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol 2006; 24: 778-89. [ Links ]
351. Mahnert B, Tauber S, Kriegmair M, Nagel D, Holdenrieder S, Hofmann K, et al. Measurements of complement factor H-related protein (BTA-TRAK assay) and nuclear matrix protein (NMP22 assay)-useful diagnostic tools in the diagnosis of urinary bladder cancer? Clin Chem Lab Med 2003; 41: 104-10. [ Links ]
352. Glas AS, Roos D, Deutekom M, Zwinderman AH, Bossuyt PM, Kurth KH. Tumor markers in the diagnosis of primary bladder cancer. A systematic review. J Urol 2003; 169: 1975-82. [ Links ]
353. Malik SN, Murphy WM. Monitoring patients for bladder neoplasms: what can be expected of urinary cytology consultations in clinical practice. Urology 1999; 54: 62-6. [ Links ]
354. van der Poel HG, Debruyne FM. Can biological markers replace cystoscopy? An update. Curr Opin Urol 2001; 11: 503-9. [ Links ]
355. Sanchez-Carbayo M, Urrutia M, Gonzalez de Buitrago JM, Navajo JA. Utility of serial urinary tumor markers to individualize intervals between cystoscopies in the monitoring of patients with bladder carcinoma. Cancer 2001; 92: 2820-8. [ Links ]
356. Lokeshwar VB, Soloway MS. Current bladder tumor tests: does their projected utility fulfill clinical necessity? J Urol 2001; 165: 1067-77. [ Links ]
357. Raitanen MP, Kaasinen E, Lukkarinen O, Kauppinen R, Viitanen J, Liukkonen T, Tammela TL. Analysis of false-positive BTA STAT test results in patients followed up for bladder cancer. Urology 2001; 57: 680-4. [ Links ]

