










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.45 no.2 La Plata abr./jun. 2011
BIOQUÍMICA CLÍNICA
Eriptosis, la muerte suicida de eritrocitos: mecanismo y enfermedades asociadas
Eryptosis, suicidal erythrocyte death: mechanism and related diseases
Eriptose, a morte suicida de eritrócitos: mecanismo e doenças associadas
Vanesa Herlax1a, Romina Vazquez2a, Sabina Mate3a, Laura Bakás4a,b
1 Dra. de la Facultad de Ciencias Exactas, UNLP.
2 Bioquímica, UNLP.
3 Dra. de la Facultad de Ciencias Exactas, UNLP.
4 Dra. en Bioquímica (orientación Bioquímica Clínica), UNLP.
a Instituto de Investigaciones Bioquímicas La Plata (INIBIOLP), CCT - La Plata, CONICET. Facultad de Ciencias Médicas. Universidad Nacional de La Plata. Calles 60 y 120, (1900) La Plata, Argentina.
b Departamento de Ciencias Biológicas. Facultad de Ciencias Exactas. Universidad Nacional de La Plata. Calles 47 y 115, (1900) La Plata, Argentina.
Resumen
La muerte suicida de eritrocitos o eriptosis, similar a la apoptosis de células nucleadas, está caracterizada por disminución del volumen celular, vesiculación de la membrana y traslocación de fosfolípidos de la membrana plasmática con exposición de fosfatidilserina en la superficie celular. Una amplia variedad de drogas, contaminantes ambientales, sustancias endógenas, condiciones clínicas y enfermedades disparan el proceso de eriptosis, entre los que se pueden enumerar cationes como Hg+2, Cd+2, sepsis, síndrome urémico hemolítico, enfermedad de Wilson y depleción de fosfato, entre otras. Este proceso es estimulado por la activación de canales iónicos y la formación de ceramida, desencadenando la activación de una compleja red de señalización. Los desencadenantes del proceso de eriptosis, así como las moléculas involucradas en la señalización del mismo, estarían también involucrados en la regulación de la apoptosis, por lo que en ambos casos, la ruta de transducción de señales involucradas serían similares. Es por eso, que los resultados obtenidos del análisis del proceso de eriptosis podrían ser potencialmente tomados como modelo en el estudio de la patogénesis de la muerte suicida de células nucleadas.
Palabras clave: Eriptosis; Muerte celular programada; Fosfatidilserina; Esfingomielinasa; Calcio intracelular
Summary
Similar to the apoptosis of nucleated cells, suicidal erythrocyte death or eryptosis is characterized by cell shrinkage, membrane blebbing and membrane phospholipid scrambling with phosphatidylserine exposure at the cell surface. A wide variety of drugs, enviromental contaminants, endogenous substances, clinical conditions and diseases trigger the eryptosis process. Examples presented include cations like Hg+2, Cd+2, sepsis, haemolytic uremic syndrome, Wilson's disease and phosphate depletion, among others. This procces is stimulated by the activation of ionic channels and the formation of ceramide, triggering the activation of a complex signaling pathway. Injuries originating eryptosis and the molecules involved in the signaling of this process may be also involved in the regulation of apoptosis, so, in both cases, the signal transduction pathway may be the same. For these reasons, the results from the analysis of eryptosis may be potentially used as a model of the pathogenesis of nucleated cells.
Key words: Eryptosis; Programmed cell death; Phosphatidylserine; Sphingomyelinase; Intracellular calcium
Resumo
A morte suicida de eritrócitos ou eriptose, similar à apoptose de células nucleadas, está caracterizada pela diminuição do volume celular, vesiculação da membrana e translocação de fosfolipídeos da membrana plasmática com exposição de fosfatidilserina na superfície celular. Uma ampla variedade de drogas, poluentes ambientais, substâncias endógenas, condições clínicas e doenças desencadeiam o processo de eriptose, entre os quais podemos enumerar cationes como Hg+2, Cd+2, sepse, síndrome hemolítico-urêmica, doença de Wilson e depleção de fosfato, dentre outras. Este processo é estimulado pela ativação de canais iônicos e a formação de ceramida, desencadeando a ativação de uma complexa rede de sinalização. Os desencadeantes do processo de eriptose bem como as moléculas envolvidas na sinalização do mesmo, estariam também envolvidos na regulação da apoptose, portanto em ambos os casos, o caminho de transdução de sinais envolvidas seria similar. É por isso que os resultados obtidos da análise do processo de eriptose poderão ser potencialmente tomados como modelo no estudo da patogênese da morte suicida de células nucleadas.
Palavras chave: Eriptose; Morte celular programada; Fosfatidilserina; Esfingomielinasa; Cálcio intracelular
Eritrocitos: modelo para el estudio de la fisiopatología celular
Los eritrocitos circulantes son las células más abundantes en un organismo adulto, constituyendo un 10% del volumen celular total. Además, son células de fácil obtención y sus funciones pueden ser estudiadas ex vivo. Esto hace que los eritrocitos sean un excelente modelo para estudiar la fisiopatología celular.
Sin embargo, estas células poseen funciones limitadas, ya que, al diferenciarse y madurar, pierden su núcleo, los ribosomas y las mitocondrias, perdiendo al mismo tiempo su capacidad de sintetizar proteínas. Producen ATP exclusivamente por degradación de glucosa. Es por todo esto que los eritrocitos han sido considerados como sacos de hemoglobina (1).
Los eritrocitos circulantes están expuestos regularmente a condiciones de estrés, como a estrés oxidativo en el pulmón y a condiciones hiperosmóticas al ingresar varias veces por hora al riñón.
Además, debido a la alta tensión de O2 en sangre arterial y al contenido de Fe, continuamente se producen dentro del eritrocito especies de oxígeno reactivas ROS, (por sus siglas en inglés Reactive Oxigen Species) tales como O2(-), H2O2, HO. Existen evidencias que indican que muchas condiciones fisiológicas y patológicas, como envejecimiento celular e inflamación, se desarrollan a través de la acción de ROS. Por esto, los eritrocitos poseen una potente acción antioxidante que consiste de rutas enzimáticas y no enzimáticas que modifican las especies altamente reactivas en intermediarios menos reactivos (2).
Los eritrocitos experimentan senescencia, resultando en la eliminación de los eritrocitos envejecidos de circulación (3). Recientemente se ha demostrado que, al igual que las células nucleadas, desarrollan un proceso de muerte suicida o eriptosis (4).
Eriptosis fisiológica: Senescencia
Los eritrocitos humanos tienen una vida media útil de aproximadamente 120 días, y como otras células, la generación y destrucción del eritrocito tiene que estar estrictamente regulada.
En condiciones fisiológicas, los eritrocitos senescentes tienen IgG autóloga unida a su superficie, favoreciendo así su fagocitosis, principalmente por células de Kupffer en el hígado. El plasma contiene anticuerpos eritrocitoespecíficos que se unen a proteínas intracelulares como actina y espectrina y a dominios intra o extracelulares de proteínas de membrana, como banda 3 (5).
La IgG se une a banda 3, específicamente a eritrocitos en estadios finales de su vida, siendo suficiente un número de aproximadamente 200 moléculas de IgG unidas al eritrocito para su fagocitosis. Sin embargo, aún no se conoce si es un agregado o la ruptura de la banda 3 la entidad reconocida por IgG. La proteólisis de banda 3 ocurre por proteasas (calpaínas) que se activan por Ca+2, el que ingresa a través de canales sensibles a cambios de volumen del eritrocito. La agregación de los productos de proteólisis de banda 3 induce un aumento en la curvatura de la membrana que desencadena el proceso de vesiculización. Por lo tanto, el proceso de envejecimiento del eritrocito va acompañado por la generación de vesículas que se desprenden de los mismos, en circulación. Cualquiera sea la hipótesis correcta, ambas explican la pérdida de la interacción entre la membrana y el citoesqueleto, con un incremento en la movilidad de banda 3 y una disminución en el número de sitios de alta afinidad por la anquirina, con la formación de neoantígenos (6).
La CD47, una proteína de membrana asociada a integrina cuya función es mediar procesos de adhesión, también está involucrada en el reconocimiento de eritrocitos envejecidos por macrófagos (7). Por otro lado, durante el envejecimiento existe una disminución del volumen celular, con un incremento en la densidad de hemoglobina en las primeras etapas y una disminución en el contenido de hemoglobina en las últimas. Estos cambios se encuentran asociados a una disminución en el contenido de colesterol y fosfolípidos, con una disminución en el área de aproximadamente 20% durante el envejecimiento (8) . Esta pérdida se correlaciona con el proceso de formación de microvesículas, las que contienen IgG unida en su superficie, exponen fosfatidilserina (PS) y contienen productos de ruptura de banda 3, las que desaparecen rápidamente de circulación al ser removidas por células de Kupffer (9).
Mecanismos moleculares involucrados
Los principales mecanismos moleculares involucrados en el inicio de la eriptosis son: la disminución de la carga energética, del poder reductor y del estrés osmótico, siendo, alguna o todas estas señales, puntos de convergencia de factores que pueden provocar eriptosis.
El evento clave en los tres casos anteriormente citados es el incremento en la concentración de Ca2+ citosólico, aunque las vías por las cuales esto ocurre en cada caso sean diferentes.
En la Figura 1 se esquematizan todas las vías que se explican a continuación.
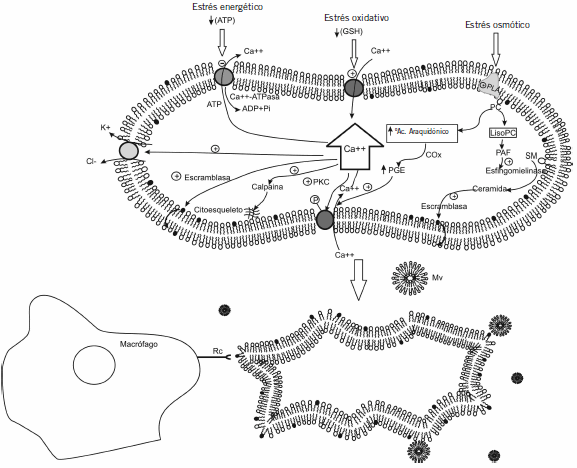
Figura 1. Esquema de las distintas vías que llevan al aumento de calcio intra eritrocitario que descencadena el proceso eriptótico.
![]() : Canales de K+ dependientes de Ca+2, canales Gardos;
: Canales de K+ dependientes de Ca+2, canales Gardos; ![]() : Canales de Ca+2 dependientes de ATP;
: Canales de Ca+2 dependientes de ATP;![]() : Canales catiónicos; GSH: glutatión reducido; PLA2: fosfolipasa A2; SM: esfingomielina; PAF: factor de agregación plaquetario; COx: ciclooxigenasa; PGE: prostaglandina E; PKC: protein quinasa dependiente de calcio; Mv: microvesículas; Rc: receptor, ≠: aumento; ↓:disminución;
: Canales catiónicos; GSH: glutatión reducido; PLA2: fosfolipasa A2; SM: esfingomielina; PAF: factor de agregación plaquetario; COx: ciclooxigenasa; PGE: prostaglandina E; PKC: protein quinasa dependiente de calcio; Mv: microvesículas; Rc: receptor, ≠: aumento; ↓:disminución; ![]() :activación;
:activación; ![]() :inhibición.
:inhibición.
En el caso de estrés energético, la reducción de ATP disminuye la actividad de la Ca2+- ATPasa y en consecuencia disminuye la salida de Ca2+ con el consiguiente incremento de Ca2+ intracelular. Esto produce la activación de PKC (proteína quinasa C, dependiente de calcio) fosforilando proteínas de membrana que permiten la entrada de Ca2+ (10).
En el estrés oxidativo, la disminución de glutatión reducido incrementa la permeabilidad a Ca2+ a través del canal de cationes, permitiendo una mayor entrada de Ca2+ al interior del eritrocito.
En cuanto al estrés osmótico, éste activa fosfolipasa A2, que libera ácido araquidónico de fosfatidilcolina. Este ácido araquidónico se convierte por acción de la ciclooxigenasa en prostaglandina E2 (PGE2), que activa los canales de Ca2+. La entrada de Ca2+ a través de canales catiónicos activaría los canales de K+ sensibles a Ca2+ (canales Gardos) presentes en la membrana del eritrocito (11), con la consecuente pérdida de KCl del eritrocito (12), favoreciendo de esta manera la hiperpolarización y la disminución del volumen celular o cell shrinkage (13).
Estos procesos disparan la vesiculización de la membrana celular (14) y estimulan la traslocación (en inglés, scrambling) de fosfolípidos de la membrana (15), produciendo la exposición de PS por medio de una escramblasa sensible a Ca+2 y/o por inhibición de una aminotraslocasa sensible a Ca+2 y dependiente de ATP (16)(17).
Un segundo estimulador de la traslocación de fosfolípidos de la membrana celular es la ceramida (18). La acción de fosfolipasa A2 genera además de ácido araquidónico, lisoderivados, los que son transformados en el factor activador plaquetario (PAF). PAF estimula a la esfingomielinasa que genera ceramida por hidrólisis de la esfingomielina (SM), capaz de estimular a la escramblasa.
El Ca2+ estimula también a la calpaína, una endopeptidasa cisteínica que degrada las proteínas del citoesqueleto y por lo tanto, inicia la vesiculización de la membrana celular (19).
El Ca2+ puede ingresar también vía canales catiónicos no selectivos (20) como TRPC6 (21) y canales catiónicos activados por ATP extracelular como P2X7 (22). Los canales son estimulados por shock hiperosmótico (23), estrés oxidativo (24) y pérdida de Cl- (23).
Impacto de los canales catiónicos en el envejecimiento de eritrocitos
Es posible especular que los canales catiónicos sensan la edad celular. Así, en los eritrocitos envejecidos, la pérdida de las defensas oxidativas incrementan la actividad de los canales catiónicos provocando un ingreso de Ca2+, aumentando la actividad de la bomba de Ca2+ y produciendo la activación de la escramblasa. Por este motivo, los eritrocitos envejecidos exponen PS en su superficie, lo que contribuye a la eliminación de estas células senescentes (25).
Todos estos hechos son también eventos típicos de la apoptosis de células nucleadas (26).
Inductores de la eriptosis
La eriptosis es un mecanismo empleado fisiológicamente por el organismo para destruir eritrocitos envejecidos sin daño necrótico, evitando de esta manera la hemólisis dentro del sistema circulatorio, lo que traería aparejado cambios similares a un proceso inflamatorio, con daños renales y alteraciones en la coagulación. Al mismo tiempo, una amplia variedad de drogas (27), contaminantes ambientales (28) y sustancias endógenas (27) pueden estimular la muerte prematura de los eritrocitos.
Los iones Hg+2 se encuentran entre los contaminantes ambientales más tóxicos. Entre las secuelas de la intoxicación crónica con Hg+2 se pueden mencionar desórdenes neuronales, anemia y falla renal (29). En cuanto al efecto sobre los eritrocitos, el Hg+2 en concentraciones micromolares, induce alteraciones en la membrana plasmática típicas de la eriptosis (30). La activación de los canales Gardos en este caso, no es debida a un incremento en la actividad de Ca+2 sino a una interacción directa con estos canales (31). En resumen, la exposición a Hg+2 lleva a una activación transiente de los canales Gardos, pérdida de K+ del eritrocito, eflujo de agua, disminución del volumen del eritrocito, activación de la esfingomielinasa y exposición de PS en la superficie del eritrocito. Efectos similares han sido encontrados para el Zn+2 en un rango de concentraciones detectadas en plasma humano (32).
La exposición de eritrocitos a Bi+3 (≥500 mM) empleado en el tratamiento de enfermedades gastrointestinales (33) o AuCl (≥0,75 μg/mL), comúnmente usado en el tratamiento de la artritis reumatoidea y contra tumores (34), produce efectos similares. Sin embargo, a diferencia de lo encontrado por efecto de Hg+2 y Bi+3, no se registró formación de ceramida por exposición de eritrocitos a AuCl (35).
En el caso de las sales de aluminio utilizadas para impedir la absorción de fosfato intestinal en la falla renal crónica, éstas causan como efecto colateral anemia como resultado de una eliminación acelerada de eritrocitos de circulación vía un proceso eriptótico. La eriptosis producida por Al+3 se produce en paralelo con la liberación de hemoglobina, demostrando que existe en este caso una pérdida de integridad de la membrana plasmática. El Al3+ causa una disminución de ATP llevando a la activación de canales catiónicos permeables a Ca2+ con el ingreso de Ca2+ a la célula, estimulación de la traslocación de PS y disminución del volumen celular. Además, se observa pérdida de hemoglobina, un evento característico de la hemólisis. El aumento de volumen (swelling) asociado con la hemólisis contrarresta la disminución de volumen (shrinkage) asociada a la eriptosis. De hecho, es posible observar una resistencia a la hemólisis hipotónica en eritrocitos tratados con Al+3. De todos modos, ambos efectos, hemólisis y eriptosis, disminuyen el tiempo de vida de los eritrocitos en circulación, contribuyendo a la anemia observada durante la intoxicación con dicho catión (36).
Inhibidores de la eriptosis
La eriptosis es inhibida por amiloride y etilisopropilamiloride (EIPA) (37) así como por catecolamina, dopamina, isoproterenol y epinefrina (38), al ser todos estos inhibidores de los canales catiónicos permeables a Ca2+.
Enfermedades asociadas a eriptosis
La eriptosis ha sido observada en una amplia variedad de condiciones clínicas o enfermedades, incluyendo síndrome urémico hemolítico (39), sepsis (40), malaria (41), anemia falciforme (42), beta talasemia (43), deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD) (43) y depleción de fosfato (44).
Síndrome urémico hemolítico (SUH)
Es una enfermedad variable caracterizada por anemia hemolítica con presencia de eritrocitos fragmentados, trombocitopenia y falla renal aguda (45). Frecuentemente el SUH es causado por la toxina del tipo Shiga, producida por Escherichia coli O157:H7 (46), y raramente por un defecto genético de las proteínas del complemento o proteasas que clivan el factor de von Willebrand (47), deficiencia del factor H (48) o por autoanticuerpos anti-factor H (49). El SUH puede también ser causado por ciertas drogas incluyendo ciclosporina, mitomicina C, anticonceptivos orales, quinina y cocaína entre otras (50).
Los experimentos presentados por Lang et al (39) muestran que el plasma de pacientes con SUH dispara la muerte suicida de los eritrocitos. Ellos demostraron que la exposición de eritrocitos a plasma de pacientes con SUH dispara el ingreso de Ca2+ y la formación de ceramida estimulando la exposición de PS, favoreciendo asi, la unión a receptores de PS expresados por macrófagos y su fagocitosis, eliminando de esta manera a los eritrocitos afectados (51).
Sepsis
Es una condición comúnmente asociada a la infección con una amplia variedad de patógenos (52). Entre las secuelas características de la sepsis se puede nombrar el desarrollo de una rápida anemia, no por una disminución en la formación de eritrocitos sino por una eliminación acelerada de los mismos de circulación (53).
Cuando eritrocitos de voluntarios sanos se exponen a plasma de pacientes sépticos o sobrenadantes de patógenos, se incrementa la exposición de PS. El efecto del plasma de pacientes sépticos sobre la concentración de Ca2+ es modesto como para ser responsable de la fuerte estimulación de la exposición de PS. En este caso, es probable que la formación de ceramida por ruptura de SM mediada por esfingomielinasa sea el factor más importante. Varias bacterias patógenas producen esfingomielinasas, que estimulan la formación de ceramida (18). Por ejemplo, la beta hemolisina de Staphylococcus aureus es uno de los agentes más frecuentes causantes de sepsis, y es una toxina que tiene actividad esfingomielinasa (54). Por otro lado, existe la posibilidad que esfingomielinasa de otro origen, como por ejemplo producida por leucocitos, sea liberada al plasma de pacientes sépticos. Independientemente del origen de la esfingomielinasa estas enzimas están críticamente involucradas en la fisiopatología de la sepsis (55).
La hemólisis inducida por determinados patógenos puede de manera similar contribuir a la eliminación acelerada de eritrocitos y producir anemia en pacientes sépticos, como se ha demostrado luego de la infección por Clostridium perfringens (52).
La ceramida (56) y el Ca2+ (57) también han sido implicados en disparar la apoptosis de células nucleadas, así como los componentes del plasma que disparan la apoptosis de células hepáticas, renales y endoteliales, contribuyendo al efecto pleiotrópico de la sepsis.
Malaria
El patógeno causante de la malaria, Plasmodium falciparum, entra en el eritrocito escapando del reconocimiento por el sistema inmune del huésped. Es por eso que Plasmodium falciparum depende de la activación de canales iónicos en el eritrocito que le permiten incorporar nutrientes, Na+ y Ca+2. A su vez, activa canales en el eritrocito por estrés oxidativo, produciendo la apertura de los canales catiónicos permeables al Ca+2 que estimulan la eriptosis (58). Mientras que los canales se requieren para la sobrevida, replicación y maduración del Plasmodium falciparum en el eritrocito, también limitan la vida del eritrocito infectado y en consecuencia del patógeno intracelular.
En conclusión, la eriptosis prematura de eritrocitos infectados antes que Plasmodium falciparum escape y propague la infección, podría ser un mecanismo protector contra la malaria, y, por lo tanto, cualquier intervención terapéutica que acelere la muerte suicida de los eritrocitos infectados podría influir favorablemente en la cura de la enfermedad.
Deficiencia de hierro
La deficiencia de hierro ocasiona anemia, la cual en parte es debida a una disminución de la vida media del eritrocito. Está aceptado que la anemia por deficiencia de hierro es el resultado de una eritropoyesis disminuida (59). Sin embargo, existen algunas evidencias que indican que la anemia en este caso también es ocasionada por una vida media disminuida de los eritrocitos deficientes en hierro.
El contenido reducido de hemoglobina en eritrocitos deficientes en hierro lleva a una disminución de la presión osmótica coloidal, y por lo tanto, a una reducción del volumen celular (60). Esta disminución de volumen es responsable de un aumento en la actividad de los canales catiónicos no selectivos (23). Aunque el ingreso de Na+ a través de este canal produce un incremento de volumen, la entrada de Ca+2 ejerce un efecto opuesto. El ingreso de Ca+2 y la activación del canal Gardos contribuyen a la activación de la eriptosis. Además, dado que los eritrocitos deficientes en hierro son más sensibles al estrés oxidativo, esto induce la activación de canales catiónicos.
Apoptosis vs. Eriptosis
La apoptosis, o muerte celular programada, es un mecanismo fisiológico que elimina las células potencialmente perjudiciales. Está desencadenada por un conjunto de reacciones bioquímicas, genéticamente reguladas, que ocurren en las células de un organismo pluricelular, encaminadas a producir la muerte de la célula de manera controlada, sin dar lugar a una reacción inflamatoria que afecte a las células vecinas. Eventos típicos de la apoptosis incluyen condensación nuclear, fragmentación de ADN, ruptura de las membranas nucleares, depolarización mitocondrial, disminución de volumen celular o shrinkage, colapso del citoesqueleto y pérdida de la asimetría de la membrana plasmática con externalización de PS (26). La exposición de PS en la superficie celular facilita la fagocitosis de las células muertas (61), produciendo la eliminación de células sin la liberación de proteínas, las cuales causan inflamación (62). Los cambios bioquímicos asociados a estos cambios morfológicos están relacionados con un incremento en la actividad endonucleasa y proteasa. Las proteínas de la familia de las caspasas necesitan ser activadas por clivaje proteolítico. Durante la apoptosis, las caspasas funcionan como iniciadores (caspasa 8 y 9) en respuesta a señales proapoptóticas o como efectores (caspasa 3) la que a través del clivaje de varias proteínas vitales induce el fenotipo apoptótico descripto arriba.
La activación de las caspasas es disparada por dos rutas de señalización. La inducción via receptores de muerte como CD95 se inicia por oligomerización del receptor por su ligando, reclutando la proteína FADD (Fas-associated death domain) al complejo, que de esta manera se activa y se asocia con la procaspasa 8, que como consecuencia se activa por autoproteólisis. La otra ruta es disparada por una serie de estímulos apoptóticos, como drogas anticancerígenas o irradiación, y se inicia por la liberación de citocromo c de la mitocondria. La liberación de citocromo c está bajo control de las proteínas pro y antiapoptóticas de la familia Bcl2. Una vez liberado el citocromo c, se asocia con Apaf1, otra molécula adaptadora que se une y activa a la procaspasa 9 en presencia de dATP. Ambas rutas convergen en la activación secuencial del efector de caspasas. Aunque las caspasas son los principales ejecutores, existen evidencias que otras proteasas denominadas calpaínas pueden estar involucradas. Estas proteínas heterodiméricas existen también como proenzimas inactivas, cuya actividad puede ser controlada por un inhibidor endógeno, calpastaína. Es interesante que varios sustratos de las caspasas sean clivados por la calpaína, incluyendo proteínas estructurales o proteínas involucradas en la transducción de señales como Bax, proteínas quinasas, etc. Las caspasas y la calpaína interactúan una con otra resultando en una mutua activación.
Una amplia variedad de estímulos inducen apoptosis, incluyendo óxido nítrico (63), radiación UV (64)(65) exposición a patógenos (66), shock ósmotico (67)(68), y la activación de receptores tales como CD95 (69), TNFα (70) y somatostatina (71).
A pesar de la falta de núcleo y mitocondrias, organelas involucradas en la apoptosis de células nucleadas, los eritrocitos expuestos al iónoforo de Ca2+ ionomicina experimentan disminución de volumen, vesiculización o blebbing de membrana y pérdida de la asimetría de la membrana plasmática con exposición de PS en la superficie, todos eventos típicos de la apoptosis de células nucleadas. Sin embargo, los eritrocitos de mamíferos en etapa fetal son nucleados, al igual que los eritroblastos. La maduración de los eritroblastos podría ser considerada, entonces, como un mecanismo de apoptosis parcial que termina con la diferenciación en eritrocitos como se esquematiza en la Figura 2. La eritropoyetina, hormona que como se sabe induce la eritropoyesis, detiene la apoptosis de los eritroblastos una vez que han comenzado su maduración, a través de una modulación positiva de la proteína antiapoptótica Bcl-XL y una disminución de la actividad de caspasas. La maduración de los eritrocitos requiere de la activación de caspasas 3, 6 y 7, involucradas en la desaparición de determinados organoides durante la maduración de los eritrocitos, así como también de ADNasas para el proceso de enucleación. Se ha informado que estas ADNasas II del eritroblasto provienen de macrófagos por un proceso llamado remoción heterofágica (1). Tanto el ingreso de Ca2+ como la subsequente activación de la escramblasa, son inhibidos por eritropoyetina (72) incrementando así la vida de los eritrocitos en circulación.

Figura 2. Por tratarse de una célula nucleada, la maduración del eritroblasto a eritrocito se produce mediante un proceso apoptótico. La eritropoyetina inactiva a Bcl-XL, proteína antiapoptótica e inhibe a las caspasas, interrumpiendo así el proceso apoptótico y alcanzando la célula el estadío de eritrocito maduro. Una vez finalizado su ciclo celular se descencadena el proceso de eriptosis, tal como se describió en la Figura 1.
Las etapas más avanzadas de la eriptosis se identifican claramente con la apoptosis, como la pérdida de potasio intracelular, la activación de canales de calcio, la activación de escramblasas, la contracción celular, la activación de esfingomielinasa y la externalización de PS, eventos que no se presentan en el eritroblasto pero sí son mecanismos necesarios para la destrucción no inflamatoria con participación de células del sistema retículo endotelial.
Sin embargo, en contraste con las células nucleadas, la eriptosis ocurre en ausencia de fragmentación de ADN y despolarización mitocondrial, aunque estos fenómenos tienen lugar en el eritroblasto durante el proceso que lleva a la producción de eritrocitos. Los eritrocitos maduros contienen cantidades considerables de caspasa-3 y caspasa-8, mientras que componentes esenciales de la cascada apoptótica mitocondrial como caspasa-9, Apaf-1 y citocromo c están ausentes. Sin embargo, las caspasas del eritrocito no se transforman en sus formas activas ni por envejecimiento ni por acción de estímulos apoptóticos, aunque es posible que la caspasa 3 y 8 participen en disfunciones del eritrocito incluyendo envejecimiento, anemias o infecciones como malaria. En su lugar, luego del incremento de Ca+2 citosólico, las calpaínas participan en las alteraciones morfológicas observadas.
Por lo expuesto, se puede decir que el eritrocito sufre una apoptosis parcial para diferenciarse desde eritroblasto, apoptosis que es detenida por eritropoyetina. Una vez que ha finalizado su vida útil tiene lugar la eriptosis con eventos similares a los que ocurren en las etapas finales de la apoptosis de células nucleadas. De esta manera los eritrocitos o eritroblastos pueden servir como células modelo de la patogénesis de la muerte suicida de células nucleadas, dependiendo del estadío de apoptosis que se quiera estudiar.
AGRADECIMIENTOS
Los autores agradecen por los diseños gráficos a Ana Bernasconi. LSB es miembro de la Carrera del Investigador CICPBA, Argentina; VH es miembro de la Carrera del Investigador, Consejo Nacional de Investigaciones Científicas y Tecnológicas (CONICET), Argentina; SM es miembro de la Carrera del Investigador CICPBA, Argentina; y RV es becaria de ANPCyT-Argentina.
CORRESPONDENCIA
DRA. LAURA BAKÁS
INIBIOLP, Facultad de Ciencias Médicas
Calles 60 y 120 (1900) La Plata, Argentina
FAX 54-221-4258988, Teléfono 54-221-482-4894
E-mail: lbakas@biol.unlp.edu.a
1. Daugas E, Cande C, Kroemer G. Erythrocytes: death of a mummy. Cell Death Differ 2001; 8: 1131-3. [ Links ]
2. Cimen M. Free radical metabolism in human erythrocytes. Clin Chim Acta 2008; 390: 1-11. [ Links ]
3. Bosman G, Willekens F, Werre JM. Erythrocyte aging: a more than superficial resemblance to apoptosis? . Cell Physiol Biochem 2005; 16: 1-8. [ Links ]
4. Lang K, Lang P, Bauer C, Duranton C, Wieder T, Huber S, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem 2005; 15: 195-202. [ Links ]
5. Kay M. Band 3 and its alterations in health and disease. Cell Mol Biol 2004; 50: 117-38. [ Links ]
6. Bosman G, Kay M. Erythrocyte aging: a comparison of model systems for simulating cellular aging in vitro. Blood Cells 1988; 14: 19-35. [ Links ]
7. Head D, Lee Z, Swallah M, Avent N. Ligation of CD47 mediates phosphatidylserine expression on erythrocytes and a concomitant loss of viability in vitro. Br J Haematol 2005; 130: 788-90. [ Links ]
8. Werre JM, Willekens FL, Bosch FH, de Haans LD, van der Vegt SG, van den Bos AG, et al. The red cell revisited: matters of life and death. Cell Mol Biol 2004; 50: 139-45. [ Links ]
9. Willekens F, Werre J, Groenen-Döpp Y, Roerdinkholder- Stoelwinder B, de Pauw B, Bosman G. Erythrocyte vesiculation: a self-protective mechanism? J Haematol 2008; 141: 549-56. [ Links ]
10. Klarl BA, Lang PA, Kempe DS, Niemoeller OM, Akel A, Sobiesiak M, et al. Protein kinase C mediates erythrocyte "programmed cell death" following glucose depletion. Am J Physiol Cell Physiol 2006; 290: C244-53. [ Links ]
11. Hoffman J, Joiner W, Nehrke K, Potapova O, Foye K, Wickrema A. The hSK4 (KCNN4) isoform is the Ca+2+-activated K+ channel (Gardos channel) in human red blood cells. Proc Natl Acad Sci USA 2003; 100: 7366-71. [ Links ]
12. Grygorczyk R, Schwarz W. Properties of the Ca+2 activated K+ conductance of human red cells as revealed by the patch-clamp technique. Cell Calcium 1983; 4: 499-510. [ Links ]
13. Maher A, Kuchel P. The Gardos channel: a review of the Ca+2 activated K+ channel in human erythrocytes. Int J Biochem Cell Biol 2003; 35: 1182-97. [ Links ]
14. Allan D, Michell R. Calcium ion dependent diacylglycerol accumulation in erythrocytes is associated with microvesiculation but not with efflux of potassium ions. Biochem J 1977; 166: 495-49. [ Links ]
15. Akel A, Hermle T, Niemoeller OM, Kempe DS, Lang PA, Attanasio P, et al. Stimulation of erythrocyte phosphatidylserine exposure by chlorpromazine. Eur J Pharmacol 2006; 532: 11-7. [ Links ]
16. Woon L, Holland J, Kable E, Roufogalis B. Ca+2 sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium 1999; 25: 313-20. [ Links ]
17. Zhou Q, Zhao J, Wiedmer T, Sims P. Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood 2002; 99: 4030-8. [ Links ]
18. Lang KS, Myssina S, Brand V, Sandu C, Lang PA, Berchtold S et al. Involvement of ceramide inhyperosmotic shock-induced death oferythrocytes. Cell Death Differ 2004; 11: 231-43. [ Links ]
19. Pant H, Virmani M, Gallant P. Calcium induced proteolysis of spectrin and band 3 protein in rat erythrocyte membranes. Biochem Biophys Res Commun 1983; 117: 372-7. [ Links ]
20. Kaestner L, Bernhardt I. Ion channels in the human red blood cell membrane: their further investigation and physiological relevance. Bioelectrochemistry 2002; 55: 71-4. [ Links ]
21. Foller M, Kasinathan RS, Koka S, Lang C, Shumilina E, Birnbaumer L, et al. TRPC6 contributes to the Ca(2+) leak of human erythrocytes. Cell Physiol Biochem 2008; 21: 183-92. [ Links ]
22. Sluyter R, Shemon AN, Hughes WE, Stevenson RO, Georgiou JG, Eslick GD, et al. Canine erythrocytes express the P2X7 receptor: greatly increased function compared with human erythrocytes. Am J Physiol Regul Integr Comp Physiol 2007; 293: R2090. [ Links ]
23. Huber S, Gamper N, Lang F. Chloride conductance and volume- regulatory nonselective cation conductance in human red blood cell ghosts. Pflugers Arch 2001; 441: 551-8. [ Links ]
24. Lang KS, Duranton C, Poehlmann H, Myssina S, Bauer C, Lang F. et al. Cation channels trigger apoptotic death of erythrocytes. Cell Death Differentiation 2003; 10: 249-56. [ Links ]
25. Boas F, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci USA 1998; 95: 3077-81. [ Links ]
26. Gulbins E, Jekle A, Ferlinz K, Grassme H, Lang F. Physiology of apoptosis. Am J Physiol Renal Physiol 2000; 279: F605. [ Links ]
27. Foller M, Huber S, Lang F. Erythrocyte programmed cell death. IUBMB Life 2008; 60: 661-8. [ Links ]
28. Zappulla D. Environmental stress,erythrocyte dysfunctions, inflammation,and the metabolic syndrome: adaptations to CO2 increases? J Cardiometab Syndr 2008; 3: 30-4. [ Links ]
29. Zalups R. Molecular interactions with mercury in the kidney. Pharmacol Rev 2000; 52: 113-43. [ Links ]
30. Eisele K, Lang PA, Kempe DS, Klarl BA, Niemöller O, Wieder T, et al. Stimulation of erythrocyte phosphatidylserine exposure by mercury ions. Toxicol Appl Pharmacol 2006; 210: 116-22. [ Links ]
31. Moschèn I, Schweizer K, Wagner C, Geis-Gerstorfer J, Lang F. Effects of gallium and mercury ions on transport systems. J Dent Res 2001; 80: 1753-7. [ Links ]
32. Kiedaisch V, Akel A, Niemoeller O, Wieder T, Lang F. Zinc-induced suicidal erythrocyte death1-3. Am J Clin Nutr 2008; 87: 1530-4. [ Links ]
33. Braun M, Föller M, Gulbins E, Lang F. Eryptosis triggered by bismuth. Biometals 2009; 22: 453-60. [ Links ]
34. Sopjani M, Foller M, Lang F. Gold stimulates Ca+2 entry into and subsequent suicidal death of erythrocytes. Toxicology 2008; 244: 271-9. [ Links ]
35. Sopjani M, Föller M, Dreischer P, Lang F. Stimulation of eryptosis by cadmium ions. Cell Physiol Biochem 2008; 22: 245-52. [ Links ]
36. Niemoeller O, Kiedaisch V, Dreischer P, Wieder T, Lang F. Stimulation of eryptosis by aluminium ions. Toxicol Appl Pharmacol 2006; 217: 168-75. [ Links ]
37. Lang KS, Myssina S, Tanneur V, Wieder T, Huber SM, Lang F et al. Inhibition of erythrocyte cation channels and apoptosis by ethylisopropylamiloride. Naunyn Schmiedebergs Arch Pharmacol 2003; 367: 391-6. [ Links ]
38. Lang PA, Kempe DS, Akel A, Klarl BA, Eisele K, Podolski M et al. Inhibition of erythrocyte "apoptosis" by catecholamines. Naunyn Schmiedebergs Arch Pharmacol 2005; 372 (3): 228-35. [ Links ]
39. Lang PA, Beringer O, Nicolay JP, Amon O, Kempe DS, Hermle T,et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J Mol Med 2006; 84: 378-88. [ Links ]
40. Kempe DS, Akel A, Lang PA, Hermle T, Biswas R, Muresanu J, et al.. Suicidal erythrocyte death in sepsis. J Mol Med 2007; 85: 269-77. [ Links ]
41. Brand VB, Sandu CD, Duranton C, Tanneur V, Lang KS, Huber SM, et al. Dependence of Plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell Physiol Biochem 2003; 13: 347-56. [ Links ]
42. Wood B, Gibson D, Tait J. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical associations. Blood 1996; 88: 1873-80. [ Links ]
43. Lang KS, Roll B, Myssina S, Schittenhelm M, Scheel-Walter HG, Kanz L, et al. Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell Physiol Biochem 2002; 12: 3665-72. [ Links ]
44. Birka C, Lang PA, Kempe DS, Hoefling L, Tanneur V, Duranton C, et al. Enhanced susceptibility to erythrocyte "apoptosis"following phosphate depletion. Pflugers Arch 2004; 448: 471-7. [ Links ]
45. Corrigan JJ, Boineau F. Hemolytic-uremic syndrome. Pediatr Rev 2001; 22: 365-9. [ Links ]
46. Bitzan M, Bickford B, Foster G. Verotoxin (shiga toxin) sensitizes renal epithelial cells to increased heme toxicity: possible implications for the hemolytic uremic syndrome. J Am Soc Nephrol 2004; 15: 2334-43. [ Links ]
47. Noris M, Remuzzi G. Genetic abnormalities of complement regulators in hemolytic uremic syndrome: how do theyaffect patient management? Nat Clin Prac Nephrol 2005; 1: 2-3. [ Links ]
48. Dragon-Durey MA, Frémeaux-Bacchi V, Loirat C, Blouin J, Niaudet P, Deschenes G, et al. Heterozygous and homozygous factor h deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol 2004; 15: 787-97. [ Links ]
49. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H et al. . Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 2005;16: 555-63. [ Links ]
50. Dlott J, Danielson C, Blue-Hnidy D, Mc Carthy LJ. Drug-induced thrombotic thrombocytopenic purpura/hemolytic uremic syndrome: a concise review. Ther Apher Dial 2004; 8: 102-11. [ Links ]
51. Fadok V, Bratton D, Rose D, Pearson A, Ezekewitz R, Henson P. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000; 405: 85-90. [ Links ]
52. Sessler C, Perry J, Varney K. Management of severe sepsis and septic shock. Curr Opin Crit Care 2004; 10: 354-63. [ Links ]
53. McArthur H, Dalal B, Kollmannsberger C. Intravascular hemolysis as a complication of Clostridium perfringens sepsis. J Clin Oncol 2006; 24: 2387-8. [ Links ]
54. Bohach G, Dinges M, Mitchell D, Ohlendorf D, Schlievert P. Exotoxins. In: Crossley KB, Archer GL (eds). The staphylococci in human disease. New York: Churchill Livingstone; 1997; 83-111. [ Links ]
55. Claus R, Bunck A, Bockmeyer C, Brunkhorst FM, Losche W, Kischerf R, et al. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FA SEB J 2005; 19: 1719-21. [ Links ]
56. Kolesnick R, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol 1998; 60: 643-65. [ Links ]
57. Perretti M, Solito E. Annexin 1 and neutrophil apoptosis. Biochem Soc Trans 2004; 32: 507-10. [ Links ]
58. Föller M, Bobbala D, Koka S, Huber S, Gulbins E, Lang F. Suicide for Survival - Death infected erythrocytes as a host mechanism to survive malaria. Cell Physiol Biochem 2009; 24: 133-40. [ Links ]
59. Arndt U, Kaltwasser JP, Gottschalk R, Hoelzer D, Moller B. Correction of iron-deficient erythropoiesis in the treatment of anemia of chronic disease with recombinant human erythropoietin. Ann Hematol 2005; 84: 159-66. [ Links ]
60. Schaefer R, Schaefer L. Hypochromic red blood cells and reticulocytes. Kidney Int 1999; 69: S44-8. [ Links ]
61. Eda S, Sherman I. Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell Physiol Biochem 2002; 12: 373-83. [ Links ]
62. Gulbins E, Szabo I, Baltzer K, Lang F. Ceramide induced inhibition of T lymphocyte voltage-gated potassium channel is mediated by tyrosine kinases. Proc Natl Acad Sci USA 1997; 94: 7661-6. [ Links ]
63. Ibe W, Bartels W, Lindemann S, Grosser T, Buerke M, Boissel JP, et al. Involvement of PKC and NFkappa B in nitric oxide induced apoptosis in human coronary artery smooth muscle cells. Cell Physiol Biochem 2001; 11: 231-40. [ Links ]
64. Kulms D, Poppelmann B, Yarosh D, Luger T, Krutmann J, Schwarz T. Nuclear and cell membrane effects contribute independently to the induction of apoptosis in human cells exposed to UVB radiation. Proc Natl Acad Sci USA 1999; 96: 7974-9. [ Links ]
65. Rosette C, Karin M. Ultraviolet light and osmotic stress:activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996; 274: 1194-7. [ Links ]
66. Fillon S, Lang F, Jendrossek V. Pseudomonas aeruginosa triggered apoptosis of human epithelial cells depends on the temperature during infection. Cell Physiol Biochem 2002; 12: 207-14. [ Links ]
67. Lang F, Busch G, Ritter M, Volkl, H, Waldegger, S, Haussinger D. Functional significance of cell volume regulatory mechanisms. Physiol Rev 1998; 78: 247-306. [ Links ]
68. Lang F, Madlung J, Siemen D, Ellory C, Lepple-Wienhues A, Gulbins E. The involvement of caspases in the CD95(Fas/Apo-1)- but not swelling-induced cellular taurine release from Jurkat T-lymphocytes. Pflugers Arch 2000; 440: 93-9. [ Links ]
69. Lang F, Szabo I, Lepple-Wienhues A, Siemen D, Gulbins E. Physiology of receptor-mediated lymphocyte apoptosis. News Physiol Sci 1999; 14: 194-200. [ Links ]
70. Lang K, Fillon S, Schneider D, Rammensee H, Lang F. Stimulation of TNF alpha expression by hyperosmotic stress. Pflugers Arch 2002; 443: 798-803. [ Links ]
71. Teijeiro R, Rios R, Costoya J, Castro R, Bello JL, Devesa J, et al. Activation of human somatostatin receptor 2 promotes apoptosis through a mechanism that is independent from induction of p53. Cell Physiol Biochem 2002; 12: 31-8. [ Links ]
72. Quintanar Escorza M, Calderon Salinas J. Eriptosis: La apoptosis del eritrocito. Revista Educ Bioquim (Universidad Autónoma de México) 2006; 25: 85-9. [ Links ]
Aceptado para su publicación el 21 de diciembre de 2010

