










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.47 no.3 La Plata set. 2013
BIOLOGÍA MOLECULAR
Hallazgo de patrones para péptidos-vacuna con capacidad de acople universal en moléculas de HLA-II
Identification of patterns for peptide-vaccines in universal capacity coupling HLA-II molecules
Identificagáo de padroes para peptídeos-vacina com capacidade de acoplamento universal em moléculas HLA-II
Adrián Cortés1a, Jonathan Coral1b, Ricardo Benítez Benítez2b
1 Químico.
2 Magíster en Ciencias Químicas. Ph. D. Profesor Titular.
a Instituto de Investigación en Vacunas Sintéticas, Antisuero y Nuevos Medicamentos IVSI; CP 190002, 3006234641; Popayán - Cauca, Colombia.
b Grupo de Investigación en Química de Productos Naturales, Departamento de Química, Universidad del Cauca. CP 190002, 8209800 Ext. 2334; Popayán - Cauca, Colombia
Resumen
Partiendo del alineamiento múltiple de secuencias proteicas humanas obtenidas de las bases de datos del National Center of Biotechnology Information (NCBI) y su posterior análisis espacial tridimensional, se estableció la existencia de un patrón de acople universal para péptidos presentados por las moléculas de histocompatibilidad HLA-II (DR, DP y DQ), siendo una base para el diseño de vacunas proteicas. Estos patrones espaciales fueron claramente exhibidos por los residuos altamente conservados de los tres tipos de moléculas de HLA-II. La aplicación de este nuevo hallazgo permitió diseñar péptidos con mejores valores de acople péptido-HLA-II, que los generados por el péptido de acople universal conocido como CLIP (class Il-associated invariant chain peptide).
Palabras clave: alineamiento múltiple * Complejo Mayor de Histocompatibilidad Clase II * péptido asociado a la cadena invariante del Complejo Mayor de Histocompatibilidad Clase II * antígenos leucocitarios humanos tipo II * diseño de vacunas
Summary
Starting from the multiple alignment of human protein sequences obtained from the NCBI database (National Center of Biotechnology Information) and subsequent three-dimensional spatial analysis, the existence of a pattern of universal coupling to peptides presented by MHC molecules HLA-II (DR, DP and DQ) was established, being a basis for the design of protein vaccines. These spatial patterns were clearly exhibited by highly conserved residues of the three kinds of HLA-II molecules. The application of this new finding made it possible to design peptides with better Peptide -HLA-II coupling values than those generated by the universal coupling peptide called CLIP (class II-associated invariant chain peptide).
Key words: multiple alignment * Major Histocompatibility Complex Class II * peptide invariant chain associated with Major Histocompatibility Complex Class II * human leukocyte antigen-type II * vaccine design
Resumo
FA partir do alinhamento múltiplo de sequéncias de proteínas humanas obtidas a partir das bases de dados do NCBI (National Center of Biotechnology Information) e análise espacial tridimensional subsequente, estabeleceu-se a existéncia de um padráo de acoplamento universal para peptídeos apresentados pelas moléculas de histocompatibilidade HLA-II (DR, DP e DQ), sendo uma base para o desenho de vacinas proteicas. Estes padroes espaciais foram claramente exibidos pelos residuos altamente conservados dos trés tipos de moléculas de HLA-II. A aplicagáo deste novo achado permitiu desenhar peptídeos com melhores valores de acoplamento peptídeo-HLA-II, do que aqueles gerados pelo peptídeo de acoplamento universal conhecido como CLIP (classe II-peptídeo associado a cadeia invariante).
Palavras-chave: alinhamento múltiplo * Complexo Maior de Histocompatibilidade Classe II * peptídeo associado á cadeia invariante do Complexo Maior de Histocompatibilidade Classe II * antígenos leucocitários humanos do tipo II * desenho de vacinas
Introducción
Los tres tipos de moléculas de histocompatibilidad del HLA-II DR, DP y DQ actúan como presentadoras de antígeno a las moléculas TCR presentes en las células T-helper. Estos tres tipos de moléculas de HLA-II son altamente polimórficas pero presentan estructuras tridimensionales comunes, como la hendidura conformada por las dos cadenas proteicas A y B, entre las cuales se acoplan los péptidos de origen extracelular que serán presentados (1).
El alto polimorfismo de los tres isotipos de HLA-II humano (DR, DP y DQ) es lo que dificulta la predicción y diseño de péptidos-vacuna con un acople efectivo a estas moléculas (2-4). En el mecanismo de presentación de antígenos se han identificado tres problemas que restringen la presentación de los péptidos vacuna:
1. Garantizar la entrada del péptido-vacuna a la hendidura de la molécula de HLA-II. Este aspecto es limitado por factores de impedimento estérico, ocasionado por las cadenas laterales de los aminoácidos del antígeno presentado, además, es necesario evitar los mecanismos de repulsión electrostática generados por residuos con cargas eléctricas iguales entre el péptido presentado y las moléculas de HLA-II, lo cual inhibe el acople (5).
2. Obtener suficiente estabilidad de acople en el complejo formado entre el HLA-II y el péptido, para garantizar prolongados tiempos de presentación del antígeno-vacuna y consecuentemente una efectiva activación de memoria inmunogénica (6)(7).
3. El complejo HLA-II/péptido debe presentar un registro de enlace único, de otro modo habrá poca o ninguna activación de los receptores TCR de las células T-helper (8).
Con los hallazgos obtenidos en la presente investigación, se postula la existencia de patrones moleculares universales en las moléculas de HLA-II (DR, DP y DQ) lo cual abre la posibilidad de realizar un diseño lógico, rápido y efectivo de péptidos candidatos para vacuna contra cualquier patógeno existente.
Materiales y Métodos
Para la realización de esta investigación se analizaron 299 secuencias proteicas de HLA-II, presentes en la NCBI, base de datos del NIH (National Institutes of Health) de libre acceso.
La búsqueda de homologías para las moléculas de HLA-II (DP, DQ, DR) se ejecutó mediante comparación de alineamientos realizados con algoritmos bioinformáticos para residuos de aminoácidos altamente conservados, además de ubicarlos en las estructuras 3D de los dominios de la proteína, usando sistemas de HLA-II con péptido acoplado, mediante los programas Cn3D (visua-lizador de estructuras macromoleculares, versión 4.3, 2011; http: // www.ncbi.nlm.nih.gov/Structure/ CN3D/ cn3d.shtml) y Jmol (Visor Java de código abierto para estructuras químicas en tres dimensiones. versión 12.0, 2011; http://www.jmol.org/).
Se utilizó la herramienta de investigación de alineamiento local conocida como BLAST ( Basic Local Alignment Search Tool) (9) a fin de reconocer los dominios conservados con estructura 3D de las proteínas de histocompatibilidad clase II. El hallazgo de secuencias proteicas y estructuras tridimensionales se realizó en las bases de datos del NCBI y PDB ( Protein Data Bank) utilizando dominios de proteínas cristalo-grafiadas, encontrando los alotipos DP (MMDB: 81423 / PDB: 3LQZ), DQ (MMDB: 51735 / PDB: 1UVQ) y DR (MMDB: 764 /PDB: 1DLH), los cuales fueron elegidos por presentar complejos HLA-II/péptido, y los complejos proteicos HLA-II-DR/Péptido/TCR (MMDB:88951 /PDB: 3O6F). Se depuraron estos bancos eligiendo únicamente secuencias humanas. Con la información ya seleccionada, se realizó la búsqueda de los residuos altamente conservados en esta base de datos mediante alineamiento múltiple con herramientas como t-coffe y Clustal-W. Los parámetros para escoger los residuos de aminoácidos fueron principalmente la conservación en diferentes secuencias que están en el banco de datos depurado, tomado del NCBI (12 secuencias mínimo y 17 secuencias máximo). Posteriormente, los residuos fueron localizados en las estructuras virtuales reportadas, mediante el uso de las herramientas Cn3D y Jmol.
La cuantificación de los valores de binding de los péptidos inferidos por la metodología usada a las moléculas de HLA-II se realizó con el algoritmo MultiRTA(10)(11), cuyos resultados fueron tabulados y graficados.
Resultados
Los resultados del Alineamiento Múltiple de secuencias proteicas obtenidos mediante las herramientas t-coffe y Clustal-W se exponen en la Figura 1, mostrando el análisis de un segmento de 50 residuos de secuencias humanas, indicándose en la última fila el grado de conservación de los residuos.

Figura 1. Reconocimiento de residuos altamente conservados, mostrando la frecuencia de aparición en el dominio MHC-II alpha de la HLA-DR1 (PDB: 1DLH). Residuos con asterisco (*) son altamente conservados.
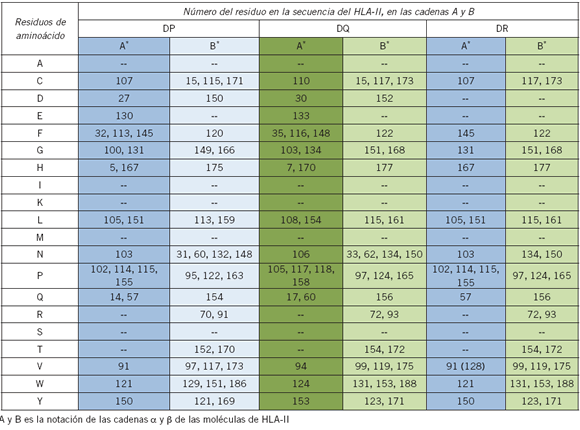
En la Tabla I se observan los residuos de alta conservación identificados en las moléculas de TCR's; por su parte, en la Tabla II se expone el número correspondiente a los residuos altamente conservados en las secuencias de los tres tipos de moléculas de HLA-II (DP, DQ y DR).
Tabla I. Residuos altamente conservados de las secuencias de TCR's en los bancos de datos del NCBI (Complejo HLA-II-DR/Péptido/TCR PDB: 3O6F)
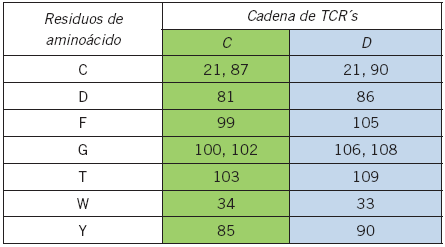
Tabla II. Residuos de las secuencias de HLA-II altamente conservados en los bancos de datos del NCBI.
En las Figuras 2 y 3 se observan alineamientos plana-res de los residuos de asparagina y de triptofano (Fig. 2) (Fig. 3) altamente conservados en las moléculas HLA-II/DP (PDB: 3LQZ) y el complejo HLA-II-DR/Péptido/TCR (PDB: 3O6F. El mismo alineamiento planar se aprecia desde otra perspectiva en las Figuras 2b y 3b, mostrando los residuos de asparagina y triptofano de alta conservación.
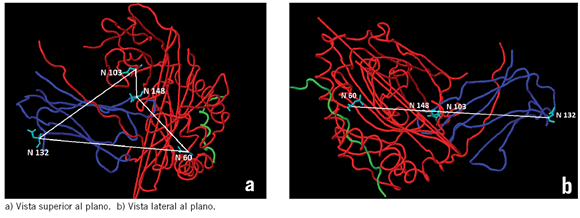
Figura 2. Posiciones espaciales de las asparaginas (N) altamente conservadas en la proteína HLA-II-DP (PDB: 3LQZ). Campo planar de asparagina (imágenes tomadas de Jmol 12.0).

Figura 3. Posiciones espaciales de triptófano (W) altamente conservados en el complejo proteico HLA DR- Péptido- TCR (PDB: 3O6F). Campo planar de triptofanos que trasciende el complejo (imágenes tomadas de Jmol 12.0).
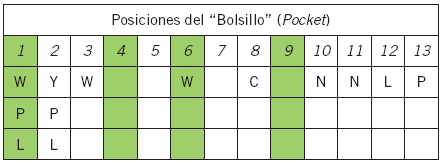
En la Tabla III se exponen las posiciones de los residuos inferidas a partir de las proyecciones planares en los "bolsillos" (pockets) de las moléculas de histocompatibilidad humanas clase II.
Tabla III. Residuos de acople universal inferidos a partir de los campos planares.
En la Tabla IV se exponen los residuos altamente conservados del HLA-II que interactúan por contacto físico directo (interacción por iguales) con los péptidos presentados (residuos F1, F3 y Q5).
Tabla IV. Residuos de acople universal inferidos a partir del fenómeno de interacción por iguales.

En la Tabla V se observan los residuos de las moléculas HLA-II que interactúan con los aminoácidos 3 y 5 de los péptidos presentados.
Tabla V. Residuos altamente conservados en HLA-II (DP, DQ y DR) que cumplen el criterio de interacción por iguales.
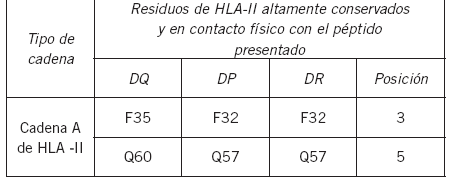
En la Tabla VI (a y b) se exponen los resultados de cuantificación del binding de los péptidos CLIP sustituidos por los residuos sugeridos en las Tablas III y IV.
Tabla VI. Predicciones de acople a moléculas de HLA-II de las secuencias CLIP sustituidas en posiciones 1, 3 y 5 por residuos F, F y Q, respectivamente; para a) tipo DR y b) tipo DP.
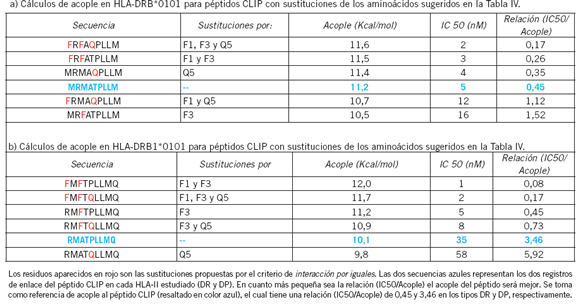
La Figura 4 representa la cuantificación del binding de los péptidos CLIP sustituidos por los residuos sugeridos en las Tablas III y IV.
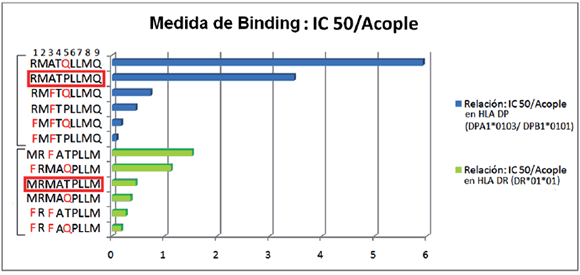
Figura 4. Medida de binding; relación IC50/Acople de los CLIP sustituidos en posiciones 1, 3 y 5 calculados con el software MultiRTA (10)(11).
La Tabla VII muestra seis péptidos diseñados fusionando los dos criterios de campos planares e interacción por iguales en un solo péptido, reuniendo los modelos de las Tablas III y IV.
Tabla VII. Medidas de binding en secuencias de CLIP sustituidos de acuerdo a los criterios de campos planares y apareamiento de iguales aplicados simultáneamente, calculados con el software MultiRTA (10)(11).
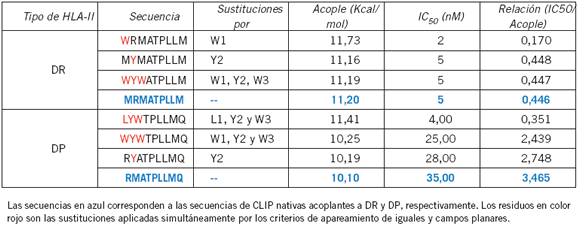
Los residuos aparecidos en rojo son las sustituciones propuestas por el criterio de interacción por iguales. Las dos secuencias azules representan los dos registros de enlace del péptido CLIP en cada HLA-II estudiado (DR y DP). En cuanto más pequeña sea la relación (IC50/Acople) el acople del péptido será mejor. Se toma como referencia de acople al péptido CLIP (resaltado en color azul), el cual tiene una relación (IC50/Acople) de 0,45 y 3,46 en los tipos DR y DP, respectivamente.
Las secuencias en azul corresponden a las secuencias de CLIP nativas acoplantes a DR y DP, respectivamente. Los residuos en color rojo son las sustituciones aplicadas simultáneamente por los criterios de apareamiento de iguales y campos planares.
Discusión
La búsqueda en dominios conservados de las secuencias de proteínas del HLA-II encontradas en el banco de datos del NCBI, condujeron a la identificación de residuos puntuales de alta conservación en los tres tipos de moléculas HLA-II de secuencias humanas. En la Figura 1 se observa un ejemplo de alineamiento múltiple del dominio MHC-II a de la proteína HLA-DR1 (MMDB: 764 /PDB: 1DHL), el cual muestra el análisis de un segmento de 50 residuos de las secuencias humanas escogidas, indicando su alta conservación al pie con un asterisco (*).
Bajo este criterio de alta conservación (columnas de letras con asterisco, como en este caso 14/14), se localizaron las posiciones de los residuos encontrados en las secuencia de las proteínas de HLA-II, arrojando los resultados de las Tablas I y II. En la Tabla I se observan los residuos altamente conservados encontrados en moléculas de TCR's; en la Tabla II se expone el número correspondiente a los residuos altamente conservados en las secuencias de los tres tipos de moléculas de HLA- II (DP, DQ y DR), tanto en sus cadenas A como B. En estos resultados se observa que las cadenas del tipo HLA-II/DR se relacionan con las otras dos moléculas DP y DQ por similitud de conservación de residuos. Esto es claro si se observa la cadena A del tipo DP (DP-A), y la cadena B del tipo DQ (DQ-B), las cuales presentan relación con las cadenas A y B del tipo DR, respectivamente. Otro aspecto que subyace en estos datos es que existen pequeñas diferencias en la posición secuencial de residuos entre las cadenas A del tipo DQ (DQ-A) y B del tipo DP (DP-B), las cuales presentan tres residuos menos en DQ-A y dos residuos menos en DP-B, con respecto a las cadenas A y B del tipo de HLA-II/DR. Al localizar espacialmente estos residuos en las estructuras 3D de cada molécula de HLA-II (DP, DR y DQ) fue posible establecer la relación en la conformación espacial de estas tres proteínas, de manera similar a lo reportado por otros estudios (12). Este patrón se aprecia en la Figuras 2 y 3.
La localización espacial de los residuos altamente conservados en cada una de las estructuras tridimensionales de las proteínas de histocompatibilidad humana clase II y TCR's, exhibieron patrones bien definidos de alineamiento en el espacio, formando un sistema de planos tal como se expone en las Figuras 2 y 3, donde se observan alineamientos planares de los residuos de asparaginas (Figura 2) y de triptófanos (Figura 3) altamente conservados en las proteínas HLA-II/DP (PDB: 3LQZ) y el complejo HLA-II-DR/Péptido/TCR (PDB:
3O6F), respectivamente. Este alineamiento planar se aprecia desde otra perspectiva en las Figuras 2b y 3b, donde una vista lateral a los planos muestra el alineamiento de las cuatro asparaginas y los seis triptofanos, respectivamente. Este fenómeno de configuración planar se presentó por igual en todos los residuos altamente conservados de las tres proteínas de HLA-II estudiadas (DR, DP y DQ). En conformidad a lo anterior, los autores de esta investigación plantean como hipótesis que el fenómeno de configuración planar entre los aminoácidos altamente conservados, se debe a la existencia de campos electromagnéticos planares que intercomunican a los residuos iguales. Tales planos se hallaron de forma recurrente en la conformación espacial de las tres clases y subclases de las moléculas de histocom-patibilidad clase II, aún a pesar del alto polimorfismo exhibido por todas estas moléculas, lo cual constituye a los planos en un patrón espacial invariable y común a todas ellas.
Este fenómeno de arreglos planares entre los aminoácidos altamente conservados hallados en esta investigación puede compararse análogamente con los planos macroscópicos exhibidos por las galaxias (20), donde este tipo de arreglos se deben a los efectos de largo alcance ocasionado por las fuerzas gravitaciona-les, mientras que en las proteínas estos arreglos podrían atribuirse a los efectos de fuerzas electromagnéticas que comunican y estabilizan residuos iguales altamente conservados. Este fenómeno de planos también quedó evidenciado al estudiar complejos proteicos completos de HLA-Péptido-TCR (PDB: 3O6F) presentados en la Figura 3, donde se observa que los campos planares trascienden a todo el complejo, mediante una alineación planar de residuos iguales (en este caso W). Este fenómeno se repite con otros aminoácidos como C y Y (resultados no mostrados), cuyo denominador común es su alta conservación en las proteínas HLA-DR y TCR respectivamente, según los bancos de datos del NCBI.
A partir de los patrones planares que se proyectan desde las moléculas de HLA-II hacia el péptido presentado, se infirió una secuencia teórica de péptido con capacidad de acople universal a los tres tipos de HLA-II (DR, DP y DQ). El péptido teórico resultante es presentado en la Tabla III, la cual muestra las posiciones y residuos que favorecen el acople en cada posición.
En la Tabla III, se observan las posiciones en los "bolsillos" (pockets) de las moléculas de histocompatibilidad humanas que favorecen el anclaje de diferentes residuos de aminoácidos en un péptido presentado según nuestro modelo de campos planares. En las posiciones 1 y 2 se aceptan residuos aromáticos como triptófano (W) y tirosina (Y), además de sustituciones por prolinas (P) y leucinas (L). Esta promiscuidad de acople de residuos en estas dos primeras posiciones es debida a la convergencia de campos planares sobre un mismo punto. Así, sobre la posición 1 convergieron campos planares de W, P, L, y sobre la posición 2 convergieron campos planares de Y, P, L. Una corroboración de estos resultados se halla en estudios experimentales realizados por otros investigadores, que muestran una alta prevalencia de los residuos W, Y, L, en estas posiciones en los tres tipos de HLA-II: DR, DP y DQ como lo exponen Castelli et al. (13), Wiesner et al. (14) y Wahlstróm et al. (15), respectivamente. Igualmente, los residuos de las posiciones 6, 8, 10, 11, 12 y 13 fueron predichos también por los patrones de planaridad del modelo desarrollado.
El patrón de residuos aminoacídicos de la Tabla III, inferido por los autores de esta investigación fue comparado con la secuencia de acople universal conocida como CLIP (Secuencia de residuos del CLIP (81-109): LPKPPKPVSKMRMATPLLMQ ALPMGALPQ (10)(11) (14)(16), la cual presentó coincidencias en el segmento (96-108) PLLMQALPMGALP en las posiciones 1, 2, 12 y 13 con los residuos prolina (P) y leucina (L) del modelo teórico encontrado. Este segmento del péptido CLIP es una secuencia alternativa de alto acople frente a moléculas del tipo DQ según Wiesner et al. (13), además de que las posiciones 1, 2, 12 y 13 con sustituciones por prolina (P) y leucina (L) son aceptadas en los tipos DP y DR según los siguientes autores: Doytchinova et al. (17), Cohen et al. (18); Dai et al. (19).
Lo anterior, respalda el patrón de residuos inferido a partir del sistema de campos planares de los residuos altamente conservados en los tres tipos de moléculas de HLA-II, permitiendo la elucidación de secuencias peptídicas de posible acople universal, lo cual será expuesto más adelante.
Según un principio de la teoría electromagnética (21)(22) el campo generado por una partícula crea un sistema de interacción a distancia inversamente proporcional al cuadrado de la distancias entre las partículas implicadas, lo cual, en este modelo de campos planares indica que la interacción por contacto físico entre residuos iguales debe generar un máximo de fuerza de interacción. A partir de esto se infiere que el contacto físico entre aminoácidos iguales es altamente favorable en términos de fuerza de acople; esto se ha denominado interacción por iguales. Los residuos altamente conservados del HLA-II con capacidad para hacer contacto físico directo con los péptidos presentados (residuos F1, F3 y Q5) son mostrados en la Tabla IV.
Los residuos de interacción por contacto directo de la Tabla IV, además de conservar su posición espacial en las moléculas de HLA-II, interactúan con los residuos 3 y 5 de los péptidos presentados, respectivamente (Tabla V).
En la Tabla V se observan los residuos de alta conservación y contacto físico directo con el péptido presentado, los cuales incrementan el binding en el complejo HLA-II/péptido, según el principio de interacción por iguales. Para verificar lo dicho, se analizó el acople de la secuencia universal CLIP con sustituciones en las posiciones sugeridas por la Tabla IV, mediante el algoritmo MultiRTA programado para cuantificar el grado de binding y su correspondiente registro de enlace en los tipos moleculares DR y DP (9)(10). Los resultados de las sustituciones realizadas sobre el péptido CLIP se presentan en la Tabla VI.
En la Tabla VI (a y b) las secuencias sustituidas se encuentran organizadas de mayor a menor binding de arriba hacia abajo. Se tomó como medida del binding la relación IC50/acople, la cual indica que a menor valor numérico mejora el acople del péptido con la molécula de HLA-II. Para un mejor análisis, los resultados de la Tabla VI se graficaron en la Figura 4.
La Figura 4 expone cómo se ven afectados los binding de los CLIP sustituidos. Las barras azules indican los resultados de acople obtenidos en el HLA-DP y las barras verdes en el tipo HLA-DR. Los resultados de medida del binding de los CLIP sustituidos se compararon con respecto al binding de las secuencias CLIP nativas (secuencias con recuadro rojo) para cada HLA-II analizado. Como se observa, la secuencia del CLIP con las sustituciones mostradas por debajo de las dos secuencias en recuadro rojo tienen mejor acople que el segmento nativo de CLIP, lo cual muestra que estos residuos de contacto físico directo estabilizan altamente la unión del péptido. Este fenómeno se puede homologar al de la formación de estructuras altamente estables como las micelas o las membranas celulares de bicapa lipídica (23).
Finalmente, seis péptidos teóricos fueron diseñados combinando los dos criterios simultáneamente, el de campos planares y el de interacción por iguales, fusionando los modelos de las Tablas III y IV, cuyos resultados de binding se exponen en la Tabla VII. Se observa que las relaciones de IC50/acople de los péptidos diseñados para HLA-II/DR, tuvieron valores similares o mejores que CLIP. Para los péptidos diseñados para HLA-II/DP se obtuvieron en todos los casos mejores valores de binding que para CLIP. Estos resultados confirman incrementos del acople péptido/HLA-II mediante la aplicación simultánea de los dos criterios mencionados en este estudio.
Conclusiones
Mediante la metodología planteada en este trabajo, se logró inferir un patrón peptídico con capacidad de acople universal a todas las moléculas de HLA-II (DR, DP, DQ), lo cual es de gran utilidad para el diseño de péptidos-vacuna o vacunas proteicas, con alta capacidad de binding. Igualmente, con estos hallazgos se diseñaron varios péptidos con mejor capacidad de acople a moléculas HLA-II que el péptido de acople universal conocido como CLIP, lo cual confirma la validez de las dos hipótesis planteadas por los autores. Finalmente, se descubrieron dos fenómenos nunca antes descritos por la ciencia de la proteómica, consistentes en la existencia de campos electromagnéticos planares en las proteínas y el apareamiento de residuos aminoacídicos iguales en las mismas macromoléculas, al cual se denomina interacción por iguales.
Se descubrieron patrones espaciales de tipo planar en los residuos altamente conservados de las moléculas de HLA-II, que determinan la capacidad de enlace de cada uno de los aminoácidos del péptido-vacuna con esta molécula, independientemente de su polimorfismo, lo cual puede ser de gran utilidad para el diseño de péptidos con fuerte acople y amplia promiscuidad de enlace en las diferentes moléculas de HLA-II.
Los campos planares mencionados trascienden a todo el complejo HLA-Péptido-TCR, indicando que estos planos cumplen con la funcionalidad adicional de provocar los acoples intermoleculares entre estas tres moléculas.
AGRADECIMIENTOS
Los autores expresan su agradecimiento al Instituto de Investigación en Vacunas Sintéticas, antisuero y Nuevos medicamentos "IVSI" y a la Universidad del Cauca por la infraestructura para el desarrollo del modelo.
CORRESPONDENCIA
Ricardo benítez benítez
Ph. D. Profesor Titular, Grupo de Investigación en Química de Productos Naturales, Departamento de Química, Universidad del Cauca. CP 190002, 8209800 Ext. 2334; POPAYÁN - CAUCA, Colombia E-mail rbenitez@unicauca.edu.co
Referencias bibliográficas
1. Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, et al. The crystal structure of a T cell receptor in complex with peptide and MHC Class II. Science 1999; 286: 1913-21. [ Links ]
2. Brown JH, Jardetzky TS, Gorga JC, Stern LJ, Urban RG, Strominger JI, et al. Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature 1993; 364: 33-9. [ Links ]
3. Jones EY, Lars F, Strominger JL, Siebold C. MCH class II proteins disease: a structural perspective. Nat Rev Immunol 2006; 6: 271-82.
4. Chicz RM, Urban RG, Gorga JC, Vignali DA, Lane WS, Strominger JL. Specifity and promiscuity among naturally procesed peptides pound to HLA-DR alleles. J Exp Med 1993; 178: 27-47. [ Links ]
5. Southwood S, Sidney J, Kondo A, del Guercio MF, Appella E, Hoffman S, et al. Several common HLA-DR types Sshare largely overlapping peptide binding repertoires. J Immunol 1998; 160: 3363-73.
6. Sato AK, Zarutskie JA, Rushe MM, Lomakin A, Natarajan SK, Sadegh-Nasseri S, et al. Determinants of the peptide-induced conformational change in the Human Class II Mayor Histocompatibility Complex Protein HLA-DR1. J Biol Chem 2000; 275: 2165-73.
7. Hammer J, Belunis C, Bolin D, Papadopoulos J, Walsky R, Higelin J, et al. High-affinity binding of short peptides to major histocompatibility complex class II molecules by anchor combinations. Proc Natl Acad Sci USA 1994; 91: 4456-60. [ Links ]
8. Rudensky AYu, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA Jr. Sequence analysis of peptides bound to CMH class II molecules. Nature 1991; 353: 622-7.
9. Altschul FS, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215: 403-10.
10. Bordner AJ, Mittelmann HD. Prediction of the binding affinities of peptides to class II MHC using regularized thermodynamic model. BMC Bioinformatics 2010; 11: 41-52. [ Links ]
11. Bordner AJ, Mittelmann HD. MultiRTA: A simple yet reliable method for predicting peptide binding affinities for multiple class II MHC allotypes. BMC Bioinformatics 2010; 11: 482-93.
12. Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 2011; 63: 325-35.
13. Castelli FA, Buhot C, Sanson A, Zarour H, Pouvelle-Moratille S, Nonn C, et al. HLA-DP4, the most frequent HLA II molecule, defines a new supertype of peptide-binding specificity. Immunol 2002; 169: 6928-34. [ Links ]
14. Wiesner M, Stepniak D, de Ru AH, Moustakis AK, Dri-jfhout JW, Papadopoulos GK, et al. Dominance of an alternative CLIP sequence in the celiac disease associated HLA-DQ2 molecule. Immunogenetics 2008; 60: 551-5.
15. Wahlström J, Dengjel J, Persson B, Duyar H, Rammensee HG, Stevanovic S, et al. Identification of HLA-DR-bound peptides presented by human bronchoalveo-lar lavage cells in sarcoidosis. J Clin Invest 2007; 117 (11): 3576-82. [ Links ]
16. Lee C, McConnell HM. A general model of invariant chain association with class II major histocompatibility complex proteins. Proc Natl Acad Sci USA 1995; 92: 8269-73.
17. Doytchinova IA, Flower DR. In silico identification of supertypes for class II MHCs. J Immunol 2005; 174: 7085-95. [ Links ]
18. Cohen WM, Pouvelle-Moratille S, Wang XF, Farci S, Munier G, Charron D, et al. Scanning the HIV genome for CD4+ T cell epitopes restricted to HLA-DP4, the most prevalent HLA class II molecule. J Immunol 2006; 176: 5401-8.
19. Dai S, Crawford F, Marrack P, Kappler JW. The structure of HLA-DR52c: Comparison to other HLA-DRB3 alleles. Proc Natl Acad Sci USA 2008; 105: 11893-7. [ Links ]
20. Kautsch SF. The nature of flat galaxies. PhD Thesis, University of Basel, Faculty of Science, 2006.
21. Cheng M, Gao H, Zhang Y, Tremel W, Chen JF, Shi F, et al. Combining magnetic field induced locomotion and supramolecular interaction to micromanipulate glass fibers: toward assembly of complex structures at meso-scale. Langmuir 2011; 27: 6559-64.
22. Ford GW, Weber WH. Electromagnetic interactions of molecules with metal surfaces. Physics Reports 1984; 113: 195-287. [ Links ]
23 Zhu Q, Vera C, Asaro RJ, Sche P, Sung LA. A Hybrid Model for erythrocyte membrane: a single unit of protein network coupled with lipid bilayer. Biophys J 2007; 93: 386-400.
Aceptado para su publicación el 19 de diciembre de 2012

