










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957
Acta bioquím. clín. latinoam. vol.49 no.1 La Plata mar. 2015
TOXICOLOGÍA
Mecanismos involucrados en el cáncer de mama por consumo de alcohol y alternativas para su prevención
Mechanisms involved in breast cancer induced by alcohol drinking and alternatives for prevention
Mecanismos envolvidos no câncer de mama por consumo de álcool e alternativas para sua prevenção
Gerardo Daniel Castro1a,b, María Eugenia Maciel2a,b, Leandro Néstor Quintans3a,b, José Alberto Castro1a,b
1 Doctor en Ciencias Químicas.
2 Doctora en Ciencia y Tecnología Mención Química.
3 Doctor en Ciencia y Tecnología Mención Química.
a Centro de Investigaciones Toxicológicas (CEITOX- UNIDEF). CITEDEF. J. B. de La Salle 4397, B1603ALO Villa Martelli, provincia de Buenos Aires, Argentina.
b Instituto de Investigación e Ingeniería Ambiental. Universidad Nacional de General San Martín (UNSAM). Av. 25 de Mayo y Francia, B1650ANQ San Martín, provincia de Buenos Aires, Argentina.
CORRESPONDENCIA DR. GERARDO D. CASTRO Centro de Investigaciones Toxicológicas (CEITOX-UNIDEF) CITEDEF Juan B. de La Salle 4397 B1603ALO VILLA MARTELLI, provincia de Buenos Aires Tel: 54-11-4709-8100 interno 1139; Fax: 54-11-4709-5911 E-mail: gcastro@citedef.gob.ar
Resumen
El consumo de alcohol se asocia con un riesgo incrementado para el cáncer de mama, aumentando linealmente incluso con un consumo moderado y con independencia del tipo de bebida alcohólica. El mecanismo está aún lejos de haberse establecido. Los estudios realizados por este laboratorio sugieren que el acetaldehído producido in situ y acumulado en el tejido mamario podría desempeñar un papel en los eventos mutacionales y de promoción del proceso carcinogénico. Estudios posteriores indicaron la producción de especies reactivas de oxígeno, acompañada de la disminución en los contenidos de vitamina E y de glutatión y de la actividad glutatión transferasa. El estrés oxidativo resultante también podría desempeñar un papel relevante en varias etapas del proceso carcinogénico. Por otra parte, está demostrado que los niveles plasmáticos de los estrógenos aumentan significativamente después del consumo de alcohol y que el riesgo de cáncer de mama inducido por beber alcohol guarda mayor relación con los tumores mamarios con receptores de estrógeno (ER)- positivo en comparación con los ER-negativos. Los estrógenos pueden producir cáncer de mama por acciones sobre el ER y también como auténticos carcinógenos químicos, como consecuencia de su oxidación que conduce a metabolitos reactivos. En este trabajo se presenta una hipótesis que integra los efectos del acetaldehído y del estrés oxidativo con los que implica un aumento de los niveles de estrógeno. Se analizan posibles acciones preventivas accesibles.
Palabras clave: Alcohol; Etanol; Acetaldehído; Radicales libres; Cáncer de mama; Estrés oxidativo; Estrógenos; Polifenoles.
Summary
Alcohol consumption is associated with an increased risk of breast cancer, increasing linearly even with a moderate consumption and irrespective of the type of alcoholic beverage. The precise mechanism is still far from being established. Studies by this laboratory suggest that acetaldehyde produced in situ and accumulated in mammary tissue because of poor detoxicating mechanisms might play a role in mutational and promotional events. Additional studies indicated the production of reactive oxygen species accompanied by decreases in vitamin E and glutathione contents and of glutathione transferase activity. The resulting oxidative stress might also play a relevant role in several stages of the carcinogenic process. Studies reported in the literature show that plasmatic levels of estrogens significantly increased after alcohol drinking and that breast cancer risk by alcohol is more related to ER-positive tumors than to ER-negative tumors. Estrogens are known to likely produce breast cancer by actions on ER and also act as chemical carcinogens as a result of their oxidation leading to reactive metabolites. In this review, a working hypothesis is introduced, integrating the effects of acetaldehyde and oxidative stress with those involving increased estrogen levels. Potential preventive actions are also analysed.
Keywords: Alcohol; Ethanol; Acetaldehyde; Free radicals; Mammary cancer; Oxidative stress; Estrogens; Polyphenols.
Resumen
O consumo de álcool está associado a um risco elevado de câncer de mama aumentando linearmente mesmo com o consumo moderado e independentemente do tipo de bebida alcoólica. O mecanismo está ainda longe de ter-se estabelecido. Estudos realizados por esse laboratório sugerem que o acetaldeído produzido in situ e acumulado no tecido mamário, poderia desempenhar um papel nos eventos mutacionais e de promoção do processo carcinogênico. Estudos posteriores indicaram a produção de espécies reativas de oxigênio, juntamente com uma diminuição nos conteúdos de vitamina E e de glutationa e da atividade glutationa transferase. O estresse oxidativo resultante também poderia desempenhar um papel importante em vários passos do processo carcinogênico. Aliás, está demonstrado que os níveis plasmáticos dos estrogênios aumentam significativamente após o consumo de álcool e que o risco de câncer de mama induzido por beber álcool guarda maior relação com os tumores mamários com receptores de estrogênio (ER)-positivo em comparação com os (ER)-negativos. Os estrogênios podem causar câncer de mama por ações sobre o ER e também como autênticos carcinógenos químicos, como resultado de sua oxidação que leva a metabólitos reativos. Neste trabalho apresenta-se uma hipótese que integra os efeitos do acetaldeído e do estresse oxidativo que envolvem um aumento nos níveis de estrogênio. Ações preventivas possíveis também são discutidas.
Palavras-chave: Álcool; Etanol; Acetaldeído; Radicais livres; Câncer de mama; Estresse oxidativo; Estrógenos; Polifenóis.
El problema del consumo creciente de bebidas alcohólicas
El efecto neto del consumo abusivo del alcohol es de aproximadamente 3,3 millones de muertes cada año, aún descontando el impacto benéfico del uso del alcohol sobre algunas enfermedades. Dicho en otras palabras, el abuso del alcohol da cuenta del 5,9% de todas las muertes a nivel global (1). En la actualidad, el hábito del consumo de alcohol comienza a edades cada vez más tempranas. Se ha demostrado que los adolescentes ingieren mayor cantidad de alcohol en menor tiempo en comparación con los adultos, siendo éste un fenómeno a escala mundial. A nivel mundial el consumo per capita de bebidas alcohólicas en el año 2010 equivalía a 6,2 litros de alcohol puro, consumido por cada persona de 15 años o más (lo cual se traduce en 13,5 gramos de alcohol puro por día). Una gran parte de este consumo (28,6%, o 1,76 litros por persona) era de fabricación casera y de producción ilegal, es decir, el alcohol no registrado. El consumo de bebidas alcohólicas de fabricación casera puede estar asociado con un mayor riesgo de sufrir daños debido a la presencia de impurezas o contaminantes desconocidos. Existe una gran variación entre los países para el consumo per capita (1).
El consumo fuerte y esporádico de alcohol (heavy episodic drinking) es otro patrón que mide el riesgo de consumo de alcohol, ya que se lo asocia con consecuencias serias de mortalidad y morbilidad. La Organización Mundial de la Salud define al heavy episodic drinking como el acto de beber por lo menos 60 gramos de alcohol puro (un litro y medio de cerveza, medio litro de vino o 150 mililitros de whisky), por lo menos una vez en los últimos siete días. A nivel mundial se detectó que aproximadamente el 11,5% de los bebedores tienen ocasión de presentar al menos un episodio de consumo en la semana, superando en número los hombres a las mujeres en una relación de cuatro a uno. Los hombres poseen un consumo de riesgo mucho más elevado en comparación con las mujeres en todas las regiones. Aún así, este fenómeno no es despreciable en la mujer (1). Aunque el uso del alcohol aumente gradualmente, los programas de tratamiento tienden a enfocarse en los hombres, desplazando las necesidades de las mujeres. El hábito de beber fue tradicionalmente una costumbre asociada al hombre y el control de dicho consumo recaía en la familia (específicamente, la esposa). Esta situación hoy se encuentra modificada, especialmente en la generación de mujeres jóvenes, que han aumentado significativamente el consumo de alcohol. Las diferencias entre los géneros se vuelven así cada vez más pequeñas (1).
Uno de los factores a tener en cuenta para entender la susceptibilidad de la mujer al consumo de bebidas alcohólicas es el biológico. La mujer posee menor actividad de enzimas gástricas que el hombre, tiene mayor proporción de grasa corporal (menor volumen de agua para su distribución) y su tolerancia sobre los síntomas característicos de la "resaca" es más baja. Respecto a las diferencias culturales, en algunos países asocian el consumo agudo y desmedido de alcohol con la demostración de masculinidad, por lo tanto, no se les permite beber a las mujeres como medida de sometimiento y evitan así la independencia de género. Sin embargo, con el movimiento feminista, las mujeres buscaron ocupar roles tradicionalmente masculinos y aumentaron el consumo de bebidas alcohólicas, con sus consecuencias adversas. En algunos casos, los problemas serios relacionados con la bebida se subestiman o se pasan por alto, por provocar mayor escándalo social (1).
Reiteradamente, las comunicaciones científicas y también los artículos periodísticos, internacionales y locales, han señalado el problema del creciente consumo de bebidas alcohólicas en Argentina, especialmente entre los jóvenes y adolescentes. El efecto perjudicial del consumo de bebidas alcohólicas en la salud de la juventud es particularmente serio si se considera que en lo que concierne a la salud reproductiva, éste es el rango etario más comprometido en ambos sexos. La salud reproductiva de la mujer no es un tema menor teniendo en cuenta el aumento alarmante del consumo de bebidas alcohólicas en un grupo etario más vinculado con la ventana de fertilidad. Los estudios epidemiológicos realizados en diferentes países, incluyendo Argentina, evidenciaron que el abuso del alcohol en la mujer es un problema de creciente interés. La incidencia aumentada del hábito de beber sobre la salud se observó particularmente en los grupos de mujeres jóvenes. En consecuencia, es de prever que el número de bebedoras adultas se incremente en las próximas décadas (1). En tal sentido, es importante considerar que debido a las diferencias en el metabolismo del alcohol, la mujer en comparación con el hombre, se encuentra frente a un riesgo mayor de consecuencias negativas asociadas con el consumo de grandes cantidades de alcohol.
Carcinógenesis por consumo de alcohol
El cáncer es la segunda causa de muerte en Argentina y en gran parte del mundo. Una proporción muy alta (entre un 80 y un 90%) de los cánceres humanos tienen su origen en factores ambientales, entendiendo por medio ambiente humano todo aquello que hace y constituye el ámbito en el cual el hombre desarrolla su actividad. Por otra parte, los hábitos constituyen el factor más importante del medio ambiente humano que incide en la generación de cáncer (la dieta, los hábitos de fumar y de consumir bebidas alcohólicas, y lo más importante, las interacciones sinérgicas entre estos factores). El consumo de bebidas alcohólicas ha sido parte de la cultura humana por siglos. Además del etanol y el agua, las bebidas alcohólicas pueden contener también una variedad de otros compuestos derivados de la fermentación, la contaminación y del uso de aditivos o sabores. Los productos laterales normales de la fermentación, al contrario que el etanol, son considerados seguros en términos generales, pero las bebidas alcohólicas pueden contener contaminantes que han sido evaluados como carcinogénicos por la Agencia Internacional para la Investigación en Cáncer (IARC), por ejemplo, N-nitrosaminas y aflatoxinas. Sin embargo, los contaminantes usualmente están en bajas concentraciones y, a través de las últimas décadas, han sido reducidos aún más, por lo menos en los países desarrollados (1). El efecto del consumo de bebidas alcohólicas sobre el riesgo de cáncer fue revisado por primera vez en las series de Monografías de IARC, en 1988 (2). En ese momento, se concluyó que había evidencia suficiente de carcinogenicidad para los cánceres de cavidad oral, faringe, laringe, esófago e hígado. Posteriormente, una gran cantidad de estudios epidemiológicos exploraron la relación entre el consumo de bebidas alcohólicas y el riesgo de cáncer en distintas localizaciones. La evidencia publicada para 27 sitios de cáncer fue revisada por un grupo de trabajo reunido en 2007 y esta revisión ha dado origen a un nuevo documento, en el cual se incorporó a la glándula mamaria como un sitio blanco de la acción carcinogénica (3).
El etanol es el ingrediente principal de las bebidas alcohólicas y el responsable de los efectos neuro-farmacológicos por los cuales estas son consumidas. Los estudios realizados en animales de experimentación no dan una evidencia clara que indique que el etanol por sí mismo pueda producir cánceres, aunque en la mayoría de los estudios el etanol fuera administrado por el lapso de la vida del animal. Podría decirse que el etanol es débil como carcinógeno y esta carcinogenicidad no se expresaría claramente en ese tiempo. El acetaldehído, el primer metabolito del etanol producido por el metabolismo oxidativo, es un potente mutágeno y carcinógeno pero generalmente se asume que en la célula este metabolito reactivo no puede alcanzar niveles suficientemente altos que pudieran plantear una amenaza carcinogénica. Como el etanol por sí no es considerado un carcinógeno, la relación del alcohol con los cánceres se ha estudiado principalmente por determinados efectos del etanol en la carcinogénesis inducida por otros compuestos carcinógenos conocidos.
Epidemiología del cáncer de mama inducido por el consumo de alcohol
El consumo de alcohol está relacionado causalmente con un riesgo mayor de padecer cánceres del tracto aero-digestivo superior, hígado, colon y recto, y mama femenino (3-6). Especialmente preocupante es el caso de la promoción de cáncer de mama debida al consumo crónico de alcohol. En efecto, el cáncer de mama es una causa de enfermedad y muerte extremadamente importante en las mujeres y el consumo de alcohol es uno de los pocos factores de riesgo modificables para este cáncer. La Agencia Internacional para la Investigación del Cáncer informó recientemente que para 2010 más de 100 estudios epidemiológicos han evaluado la asociación entre el consumo de bebidas alcohólicas y el riesgo de cáncer de mama (3). Además, el análisis combinado de los datos de 53 estudios realizados en todo el mundo mostró una relación dosis-respuesta entre el consumo de alcohol y el aumento del riesgo de cáncer de mama (5). Este estudio mostró un aumento del 9% en el riesgo por cada 10 gramos de alcohol ingeridos por día. De hecho, otros estudios epidemiológicos recientes, realizados sobre un total de 1.280.296 mujeres de edad mediana en el Reino Unido demostraron que aún las mujeres que consumen un promedio de sólo 10 gramos de alcohol ("un trago") por día mostraron 12% más de riesgo de cáncer de mama (4). Además, un estudio prospectivo detallado en 254.870 mujeres, realizado en ocho países europeos, informó que el 5% del cáncer de mama de la mujer era atribuible al consumo de alcohol (7).
Desafortunadamente, hay poca información sobre un mecanismo posible para este efecto y sobre efectos moduladores positivos de factores de la dieta, si es que no se evita el consumo de alcohol. Esta cuestión fue de especial interés para motivar los estudios realizados por distintos laboratorios con el propósito de entender la patogénesis de esta relación entre cáncer de mama y el consumo de bebidas alcohólicas y para, eventualmente, sugerir medidas de prevención (8-11).
Formación de acetaldehído, su acumulación y efectos
A pesar de la importancia del problema, el mecanismo por el cual el alcohol incrementa el riesgo de cáncer de mama sigue siendo desconocido. Varias líneas de evidencia indican que el acetaldehído, un producto del metabolismo del alcohol, podría desempeñar un papel en los efectos cancerígenos relacionados con el alcohol en diferentes tejidos diana (12)(13). Los experimentos con animales han demostrado claramente que el acetaldehído es una sustancia química mutagénica y carcinogénica (14-16). Además, otros factores como el estrés oxidativo, los procesos de metilación alterados, el metabolismo anormal de la vitamina A y del ácido retinoico y los niveles de hormonas perturbados, pueden ser de especial relevancia según sea el tejido diana implicado (6)(10). Respecto al caso específico del tejido mamario, el conocimiento de las concentraciones de acetaldehído fue de particular interés para este laboratorio. En términos generales, la concentración de acetaldehído en cualquier tejido, y también en el mamario, depende de su capacidad para producirlo in situ, además de lo proveniente a través de suministro de sangre y de la capacidad del órgano dado para degradarlo (17).
Se establecieron dos vías diferentes de oxidación de etanol a acetaldehído presentes en el tejido mamario de la rata. Una de ellas está ubicada en la fracción citosólica y la otra está en los microsomas (18)(19). Ambas fueron caracterizadas de manera preliminar y mostraron ser susceptibles a los efectos inhibitorios de varias sustancias químicas presentes en los alimentos. La enzima implicada en la ruta citosólica mostró ser la xantina óxidorreductasa (XOR), debido a su susceptibilidad a los efectos inhibitorios del alopurinol y por la capacidad del proceso para producirse sólo cuando la presencia de NAD+ fue acompañada por sustratos de la forma XOR de la enzima, tal como hipoxantina, xantina, cafeína, teobromina, teofilina o 1,7-dimetilxantina (18). Por otra parte, también se sabe que durante la intoxicación alcohólica aguda hay un aumento de la degradación de purinas e hiperuricemia (20)(21). El incremento de la oferta de purinas resultantes de este proceso también podría proporcionar una cantidad extra de cofactores para la vía mediada por XOR de metabolismo del etanol a acetaldehído (y también para la generación de radicales libres) en el tejido mamario. La presencia de XOR en el tejido mamario es bien conocida (22)(23), y estudios en este laboratorio demostraron su presencia en altas cantidades en las células epiteliales del tejido mamario de rata (24). Curiosamente, la actividad de esta vía metabólica citosólica aumentó significativamente después de una exposición repetida al alcohol repetitivo a través de una dieta Lieber y De Carli durante 28 días (24). La contribución de las enzimas presentes en la fracción citosólica del tejido mamario distintas de la XOR para la activación de etanol en acetaldehído, por ejemplo, la alcohol deshidrogenasa (ADH), puede ser más limitada. Por un lado, los estudios mostraron que no se pudo detectar alguna actividad de ADH en homogenatos de tejido mamario de rata (25). Más recientemente, este laboratorio informó rastros de actividad de la ADH en la fracción citosólica de tejido mamario, que era cerca de 16 veces más pequeña que en el hígado (17). Por otro lado, Triano y et al. informaron que el tejido mamario humano contiene una clase de ADH con un potencial limitado para transformar el alcohol en acetaldehído (26). Además de la vía citosólica mamaria de la oxidación del etanol a acetaldehído que se ha descrito anteriormente, este laboratorio reportó la presencia de otra que se produce en la fracción microsomal de este tejido (19). Se estableció que la transformación enzimática implicada era dependiente de oxígeno y de NADPH, pero que el citocromo P450 no estaba implicado, ya que no era inhibida por la mezcla de CO:O2 (80:20 v/v) o por SFK 525A (19). Curiosamente, esta transformación microsomal de alcohol a acetaldehído fue inhibida fuertemente por difenileneiodonio (DPI), dietilditiocarbamato de sodio, azida de sodio, ácido nordihidroguaiarético, pero no por la dapsona, el aminotriazol o la indometacina. Estos resultados sugieren la participación en esta biotransformación de una oxidasa o una peroxidasa, pero no de la lactoperoxidasa o de la ciclooxigenasa (19). No se pudo detectar la formación de cualquiera de los radicales hidroxilo o 1-hidroxietilo en esos estudios. En el curso de los siguientes estudios realizados con ratas expuestas a una dieta estándar Lieber y De Carli durante 28 días, se observó la inducción no sólo de la vía de activación citosólica mediada por XOR sino también de la microsomal (24).
Esto último fue de particular importancia, ya que en el curso de los trabajos recientes se mostró que el efecto de aumento (inducción) no se debe a la participación del CYP2E1 después de la exposición repetida al alcohol, efecto que es conocido por la fracción microsomal hepática (27). Además, la acetona, otro inductor del metabolismo microsomal del alcohol mediado por CYP2E1 en el hígado, no logró incrementar la bioactivación de etanol ni la actividad enzimática CYP2E1 en la fracción microsomal del tejido mamario de rata (27). Para asegurar que la actividad enzimática CYP2E1 no estaba presente en el tejido mamario de rata o era muy baja, también se incluyó en los estudios la determinación de la actividad clorzoxazona hidroxilasa. Esta actividad es considerada en la literatura como una respuesta significativa a la presencia de CYP2E1 en un tejido dado (27). No fue posible detectar el metabolismo de clorzoxazona en la fracción microsomal del tejido mamario a pesar del hecho que se empleó un procedimiento particularmente sensible, donde la formación de metabolito 6-hidroxiclorzoxazona se cuantificó por HPLC con detección culombimétrica (27).
Eso excluye aún más la participación de CYP2E1 en esta vía microsomal del metabolismo del alcohol en el tejido mamario y animó a desafiar la posibilidad de que una peroxidasa o una lipoxigenasa estuviera involucrada en ese proceso de oxidación. Esta hipótesis fue acuñada inicialmente debido al efecto inhibidor potente del ácido nordihidroguayarético. Este polifenol es un inhibidor conocido de las lipoxigenasas (28)(29).También existe la posibilidad de que el efecto inhibidor potente del DPI indique la participación adicional de una enzima NADPH oxidasa, como un proveedor de peróxido de hidrógeno. Desde este punto de vista, el papel de la NADPH oxidasa sería la generación del co-sustrato necesario requerido por la lipoxigenasa para ejercer su actividad contra los xenobióticos (10). En apoyo de esta hipótesis está el hecho de que el efecto inhibidor específico de DPI sobre la NADPH oxidasa está bien establecido (30). Esta hipótesis visualiza el proceso general de la oxidación microsomal del etanol a acetaldehído en el tejido mamario de rata como un mecanismo de cooperación entre la NADPH oxidasa y la lipoxigenasa (Fig. 1).
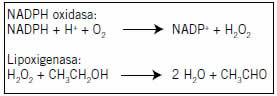
Figura 1. Mecanismo propuesto para la cooperación entre la NADPH oxidasa y la lipoxigenasa en la oxidación de etanol a acetaldehído en microsomas.
La concentración de acetaldehído en diferentes tejidos depende de la producción y de su degradación y de la cantidad del mismo que llega al tejido dado a través del suministro por la sangre. Pudo determinarse la cantidad de acetaldehído que se acumula en el tejido mamario después de dosis únicas de etanol y también los niveles en la sangre en diferentes períodos de tiempo. Los estudios se llevaron a cabo empleando tres dosis diferentes (alta, media y baja). Los valores se compararon con los que ocurren en el hígado de los mismos animales (17). Los niveles de acetaldehído en el tejido mamario fueron más altos que en el plasma y más bajos que en el hígado durante al menos 15 horas para la dosis más alta probada o seis horas para la dosis media o dos horas para el caso de la inferior. La forma de la curva de concentración de acetaldehído en el tejido mamario en función del tiempo después de la administración oral siempre imitaba a la observada en el hígado. Pero, más importante, los niveles de acetaldehído en plasma fueron similares para las tres dosis de etanol dado (17). Estos resultados sugieren que el acetaldehído presente en el tejido mamario (y en el hígado) refleja el equilibrio entre la capacidad de estos tejidos para generar acetaldehído y aquella para metabolizarlo y eliminarlo. El hígado es capaz de deshacerse del acetaldehído formado con la participación de la aldehído deshidrogenasa (ALDH) y glutatión transferasa (GST) (21). En el caso de tejido mamario la situación parece ser diferente. Por un lado, la actividad de ADH es aproximadamente 16 veces menor que en el hígado, pero tal vez más importante, la actividad ALDH presente en tres fracciones subcelulares probadas en estudios de los autores resultó en todos los casos por lo menos diez veces más baja que en el hígado (17).
La conclusión general fue que el acetaldehído es capaz de acumularse en el tejido mamario durante períodos de tiempo significativamente relevantes, principalmente como resultado de su capacidad para oxidar etanol a acetaldehído in situ y en un grado menos importante, por la llegada del mismo a través de la sangre y producido en otro lugar (por ejemplo, en el hígado) (17). ¿Cuáles podrían ser las consecuencias esperadas de la acumulación de acetaldehído en un tejido por períodos de tiempo extensos? Es importante señalar que el acetaldehído es una sustancia química reactiva que resultó ser tóxica, mutagénica y carcinogénica, y capaz de interactuar con muchos constituyentes celulares, incluyendo ADN, proteínas (proteínas nucleares), lípidos (incluyendo lípidos nucleares), glutatión y otros (21)(31)(32).
Las reacciones del acetaldehído con el ADN son de especial preocupación porque sugieren claramente que actúa como un iniciador tumoral y un compuesto mutagénico. Las estructuras de los aductos de ADN y acetaldehído identificados se revisaron recientemente en la literatura (33-35). Los aductos de ADN pueden causar errores de la polimerasa e inducir mutaciones en los genes críticos. Además, pueden dar lugar a mutaciones que activan proto-oncogenes e inactivan genes supresores de tumores en células replicantes (36)(37). No obstante, se conocen enzimas de reparación del ADN que pueden modificar ese daño del ADN causado por acetaldehído, eliminando sus aductos del ADN. La relación entre la formación de aductos y su reparación sería relevante para la epidemiología molecular del cáncer, sobre todo en las células en reproducción (37). Es de particular interés a este respecto que los estudios realizados por Freudenheim y colaboradores reportaron resultados que fueron consistentes con una mayor probabilidad de tumores con mutaciones de p53 en el cáncer de mama antes de la menopausia con el aumento de la ingesta de alcohol en los 10 ó 20 años anteriores (38). Para consumos de 16 o más tragos al mes en el período de 20 años antes de la entrevista y en comparación con los no bebedores, la OR fue de 5,25 (95 %, 1,48-18,58) (38).
Se sabe también que el acetaldehído se une a las proteínas en sus aminoácidos y el tema también podría ser relevante a los efectos carcinogénicos de beber alcohol en diferentes órganos. Sin embargo, no se dispone en la actualidad en la literatura de estudios específicos relacionados con el cáncer de mama y el consumo de alcohol y el potencial de la participación de la unión de acetaldehído a las proteínas. No obstante, se conoce que el acetaldehído se une a los residuos reactivos de lisina, a algunos aminoácidos aromáticos, a la cisteína, o a los grupos alfa-amino libres, tales como los de la valina terminal de la hemoglobina. El tema fue examinado a fondo por Niemelä, quien también señaló la importancia de esas interacciones en diferentes patologías que ocurren en varios tejidos distintos del mamario (39). En este sentido, podría ser relevante tener en cuenta que el acetaldehído se une de forma covalente a las proteínas nucleares del tejido hepático (32). En el caso del hígado, la biotransformación de etanol a acetaldehído puede producirse no sólo en el citoplasma o en el retículo endoplasmático, si no también en la membrana nuclear externa próxima (32)(40).
El acetaldehído, a pesar de ser una molécula reactiva, no tiene la reactividad equivalente a la de un radical libre de bajo peso molecular (por ejemplo, triclorometilo o 1-hidroxietilo). Esto le permite viajar desde sitios celulares distantes (citoplasma o del retículo endoplasmático) hasta el núcleo y interactuar con el ADN y las proteínas nucleares o lípidos cuando por otra parte se acumula durante largos periodos de tiempo como los observados en el tejido mamario después de beber alcohol. En el caso de otros productos químicos carcinógenos hepáticos, como el tetracloruro de carbono, se observó una buena correlación entre la unión covalente de sus metabolitos reactivos a las proteínas nucleares y la carcinogenicidad. La interacción involucraba proteínas histonas y no histonas. Las proteínas nucleares ácidas y residuales fueron los blancos favoritos de esa interacción y mostraron una buena correlación entre esas interacciones covalentes y carcinogenicidad en distintas especies (41). Además, es importante tener en cuenta que el etanol es un inductor conocido de los metabolismos mediados por el CYP2E1 (21) y que este efecto de incremento se observó en la membrana nuclear externa del hígado (42)(43).
También se pueden esperar consecuencias de largo alcance de las alteraciones en las proteínas nucleares resultantes de un ataque del acetaldehído. En efecto, se sabe que las proteínas nucleares son críticas para la división celular, el crecimiento, la diferenciación y la apoptosis (44-46). Un claro ejemplo de proteínas nucleares relevantes es aquel de las implicadas en el reloj del ciclo celular. Ellas incluyen ciclinas, quinasas dependientes de ciclina, proteínas codificadas por proto-oncogenes y genes supresores de tumores (tales como las proteínas MYC y BCL o la pRB o p53 y otras); enzimas necesarias para la síntesis de ADN y de reparación del ADN (47)(48). Diversos autores proveyeron evidencia crítica que algunas de estas actividades pueden verse afectadas por el acetaldehído producido durante el consumo de etanol, y mostraron que el consumo crónico de etanol resulta en la inhibición de la reparación de la alquilación del ADN por la O6-metilguanina transferasa (O6-MeGT), que elimina grupos alquilo de la posición O6 de la guanina. Se ha demostrado que el acetaldehído inhibe la O6-MeGT (19)(49-51). Además, se demostró en estudios recientes que la exposición a etanol interfiere con la maquinaria de la división celular por perturbar la proteína ciclina clave de G2 (52).
En cuanto a la unión covalente a lípidos nucleares, hay que señalar que en el caso del hígado, todas las fracciones de lípidos estuvieron involucradas. Parte de la unión covalente era lábil en medio ácido, pero una parte significativa no lo fue. La porción lábil podría atribuirse a la presencia de aductos del tipo base de Schiff de acetaldehído con grupos amino de fosfolípidos (32). En efecto, los estudios previos de otros laboratorios informaron que los fosfolípidos que contienen grupos amino como fosfatidilserina y fosfatidiletanolamina forman aductos de base de Schiff (53)(54). La fracción de unión covalente resistente a la hidrólisis ácida podría atribuirse en parte a las reacciones de adición de 1-hidroxietilo sobre dobles enlaces de ácidos grasos insaturados o en restos de colesterol y de los fosfolípidos que contienen nitrógeno. Ese comportamiento se había reportado anteriormente para el caso de otros radicales libres de carbono, por ejemplo los radicales triclorometilo (55)(56). Otra parte de la unión a lípidos resistente al medio ácido suave covalente también puede atribuirse a la formación de ésteres etílicos de ácidos grasos. Se sabe que los microsomas y el citosol del tejido hepático contienen sistemas de enzimas capaces de esterificar los ácidos grasos libres o transesterificar otros ésteres para generar ésteres etílicos (57-59). Si los preparados nucleares de tejido mamario tienen habilidades similares aún está por establecerse. Independientemente de la actual falta de conocimientos sobre la estructura de los productos de reacción formados, no es aventurado prever que la alteración de las membranas nucleares por su reacción con el acetaldehído o el 1-hidroxietilo como así la formación del ésteres con etanol, podría dar lugar a una profunda alteración de las funciones nucleares. Está bien establecido que el etanol también altera notablemente otras membranas hepáticas y su fluidez (60). Se sabe que los lípidos nucleares pueden ser parte de un sistema de señalización intracelular que modula la proteína quinasa C, una enzima implicada en la fosforilación de proteínas nucleares (61). La unión covalente a los fosfolípidos nucleares merece especial interés ya que ellos tienen una conocida capacidad para regular la función génica, la estructura del nucleosoma, la síntesis de ARN y la activación de la ADN polimerasa alfa (61-67).
Estudios anteriores del laboratorio reportaron correlaciones entre las alteraciones en los lípidos nucleares resultantes de un ataque de radicales libres y la respuesta diferente de las cepas de ratas utilizadas para estudiar los efectos cancerígenos de CCl4 (56). Más arriba se ha mencionado que la capacidad del tejido mamario para la oxidación del alcohol puede aumentar por la exposición repetida al mismo alcohol (24). Como se ha analizado también antes, la mayor parte de la transformación metabólica de etanol que lleva a la formación de acetaldehído está presente en la fracción citoplásmica de tejido mamario y está mediada por la XOR (10)(18)(24). Durante este proceso mediado por la XOR también se producen radicales libres hidroxilo (18). Esta actividad enzimática está presente en grandes cantidades en el tejido mamario, localizada en las células epiteliales. Además, pudo demostrarse que es inducible en el tejido mamario por la exposición repetida al alcohol y por otros compuestos de relevancia ambiental como la acetona (24)(27). Las vías metabólicas presentes en la fracción microsomal del tejido mamario también son inducibles por la exposición repetida al etanol (24). Sin embargo, este camino no dependería del CYP2E1, como pudo demostrarse (10)(27).
Por lo tanto, una consecuencia perjudicial predecible de la inducción del metabolismo del alcohol que lleva al acetaldehído y a radicales libres por la exposición repetida a bebidas alcohólicas se desprende del hecho que varios pro-carcinógenos mamarios del tipo de los nitroheterocíclicos podrían activarse por una vía nitrorreductiva mediada por la XOR (68)(69), acompañado de especies reactivas del oxígeno producidas por el ciclo redox (un aspecto que será analizado a continuación). Una situación equivalente podría plantearse para el metabolismo microsomal aumentado por la inducción del alcohol con distintos pro-carcinógenos. Si bien el análisis de los efectos potenciales del alcohol a través de la inducción del metabolismo de otros carcinógenos excede el alcance de esta revisión, es evidentemente un factor muy relevante frente a la carcinogénesis mamaria que debe tenerse en cuenta como variable.
Los radicales libres, el estrés oxidativo y sus efectos
El estrés oxidativo también se considera como un factor potencial implicado en el consumo de alcohol promoviendo un efecto cancerígeno en el tejido mamario. A pesar de esto, no había evidencia de su ocurrencia en el tejido mamario hasta los estudios recientes en este laboratorio, en los que se mostró que el estrés oxidativo se puede observar en este tejido después de un protocolo experimental de 28 días de consumo de alcohol a través de la dieta Lieber y De Carli (17)(24)(27). El estrés oxidativo se ha definido como un desequilibrio entre oxidantes y antioxidantes en favor de los primeros, dando lugar a un aumento global en los niveles celulares de especies reactivas de oxígeno (ROS). Muchos mecanismos pueden desempeñar un papel en cómo el etanol induce el estrés oxidativo en el hígado (el tema ha motivado el interés de numerosas investigaciones desde hace años) y ellos están revisados por Cederbaum y colaboradores (70). El estrés oxidativo inducido por el etanol en el hígado incluye la capacidad de generar radicales libres capaces de iniciar el proceso (por ejemplo, 1-hidroxietilo y radicales hidroxilo) y la formación de hidroperóxidos, peróxidos, anión superóxido, H2O2 y otros . En el caso del hígado expuesto al alcohol, la evidencia incluye la formación de productos derivados del ataque de ROS a moléculas diana pertinentes (por ejemplo, ADN, proteínas o lípidos) como carbonilos en proteínas, 8-oxodesoxiguanosina, la disminución de sulfhidrilos de proteínas, los hidroperóxidos de lípidos y otros productos tales como el malondialdehído o el 4-hidroxi-2-nonenal (4HNE) (70).
La evidencia de que las defensas antioxidantes celulares fueron excedidas podría incluir determinaciones de la variedad de mecanismos enzimáticos y no enzimáticos que han evolucionado para proteger las células contra las ROS, tales como la superóxido dismutasa, la catalasa, la glutatión peroxidasa, la GST y otras enzimas. También son relevantes los antioxidantes de peso molecular bajo, tales como el glutatión en sí, la vitamina E, el ascorbato, la vitamina A y otros. En resumen, la toxicidad inducida por ROS refleja el equilibrio entre las tasas de producción de ROS en comparación con las tasas de eliminación de ROS, más la reparación del daño a macromoléculas celulares (70). Después de estas consideraciones aprendidas de los mecanismos de daño celular inducido por radicales libres y de los efectos del alcohol sobre el hígado, corresponde analizar las pruebas obtenidas para el caso del tejido mamario. Inicialmente debería considerarse la posibilidad de que durante el consumo de alcohol pueda ocurrir un proceso de estrés oxidativo en el tejido mamario, ya que pudo demostrase que la exposición repetida al alcohol da lugar a un aumento de las actividades de XOR y lipoxigenasa en el tejido mamario de la rata (24). En efecto, el aumento de XOR y actividades de la lipoxigenasa por sí mismos deberían conducir no sólo a una mayor generación de ROS, sino también cuando se produce en presencia de etanol, a una mayor generación de radicales libres. Se detectó la formación de radicales hidroxilo durante el metabolismo citosólico del alcohol mediado por XOR (18).
Otro motivo que lleva a un efecto similar potencial se deriva de la observación de que el acetaldehído se acumula en el tejido mamario durante el consumo de alcohol repetitivo (17). Es bien sabido que la generación de aumento del nivel de acetaldehído puede provocar disminuciones en GSH, como se observó en el hígado, respectivamente(70). Además, GSH y enzimas GSH-dependientes, como la glutatión peroxidasa, glutatión reductasa y glutatión transferasa son una importante primera línea de defensa contra las condiciones de estrés oxidativo (70). La observación inicial que indica que el consumo de alcohol podría provocar estrés oxidativo se deriva de estudios donde se verificó un incremento de la quimioluminiscencia inducida por el t-butilhidroperóxido en homogeneizados de tejido mamario de ratas expuestas repetidamente al alcohol, en comparación con los de los animales de control (17). Las muestras de animales tratados con alcohol tuvieron una respuesta completamente diferente frente al desafío del t-butilhidroperóxido y la forma de su curva de respuesta en comparación con la observada en las muestras de control sugirieron claramente que los animales expuestos a alcohol habían disminuido sus defensas contra el desafío oxidante (17). Sin embargo, ese experimento no dio una indicación sobre la naturaleza del proceso de defensa que se veía comprometido. Además, no mostró la molécula diana que participa del proceso oxidativo que se estaba produciendo. Para responder a estas preguntas, se realizaron otros estudios que evidenciaron que después de beber alcohol repetidamente, los niveles de hidroperóxidos de lípidos en el tejido mamario se incrementaron significativamente. Además, el contenido de sulfhidrilos proteicos y de vitamina E disminuyó en los animales tratados con alcohol. Sin embargo, en estos experimentos no se observó aumento de carbonilos en proteínas o un aumento de la formación de 8-hidroxiguanina (24). Una de las razones para explicar esa respuesta diferente al estrés oxidativo provocado por el alcohol podría ser que fuera necesaria una exposición más prolongada al alcohol para alterar estos parámetros. Es decir, en este trabajo se detectaron manifestaciones tempranas de estrés oxidativo inducido por el consumo de alcohol en el tejido mamario. Si ese fuera el caso, después de períodos más prolongados de consumo de alcohol, otros efectos pertinentes podrían ocurrir, derivados de la generación de aldehídos reactivos y nocivos a partir de la peroxidación de lípidos, como el malondialdehído y el 4-hidroxi-2-nonenal. Ellos son capaces de reaccionar con las proteínas, los lípidos y las moléculas que contienen grupos sulfhidrilo (35)(70).
La reacción con los grupos sulfhidrilo que se observó podría ser de importancia teniendo en cuenta que muchas enzimas que contienen sulfhidrilo juegan un papel clave en el funcionamiento de la célula y también en los procesos de señalización celular (71-74). Los radicales libres no sólo son relevantes en relación con la etapa de iniciación de la carcinogénesis. El progreso del cáncer de mama humano a la etapa de metástasis fue vinculado con el daño del ADN inducido por radicales hidroxilo (75). Estos resultados deben ser de alguna importancia en vista de la correlación establecida entre el estrés oxidativo y la promoción de tumores y cáncer (76-78).
Aumento de los niveles de estrógenos y sus consecuencias
El aumento de los niveles plasmáticos de estrógenos por el consumo de alcohol se ha demostrado claramente en estudios de alimentación controlados en mujeres voluntarias (79-88). Además, la hipótesis sobre el papel de los estrógenos en el cáncer de mama inducido por beber alcohol está fuertemente apoyada por el hecho de que su mayor riesgo estaba relacionado con los tumores mamarios con receptores de estrógeno (ER)- positivo en comparación con los ER-negativos (89-92). Sin embargo, los estrógenos pueden ejercer sus efectos cancerígenos en el tejido mamario no sólo a través de ER, sino también por el daño directo al ADN (93). La activación metabólica de los estrógenos para generar catecol-3,4-quinonas es un ejemplo. Estos metabolitos reactivos son capaces de interactuar con el ADN para producir eventos mutacionales (94-97). Estos mayores niveles de estrógeno surgirían después de beber, porque el alcohol incrementa la actividad de la aromatasa y esto conduce a una mayor conversión de la testosterona en estrógeno, lo que resulta en una disminución de la testosterona y en el aumento del nivel de estrógenos (98). Estas consideraciones podrían ser de particular importancia, ya que es clara la evidencia en la literatura que los niveles de estrógenos altos en la sangre están asociados con un riesgo mayor de cáncer de mama (98). Un mecanismo principal de acción de los estrógenos es aquel por el cual el estrógeno estimula la proliferación celular a través de vías de señalización mediadas por ER, lo que resulta en un mayor riesgo de mutaciones durante replicaciones del ADN (99-101).
Otro camino implica el metabolismo de los estrógenos, que está mediado por el citocromo P450 1B1 y que genera 2- y 4-catecol estrógenos que a su vez pueden oxidarse fácilmente a las respectivas quinonas, incluso sin la necesidad de la catálisis enzimática o a través de iones de metales de transición (102). En este punto es especialmente importante hacer mención que el consumo de alcohol es capaz de aumentar de forma significativa el metabolismo oxidativo dependiente de NADPH de los estrógenos para dar quinonas en las hembras. El CYP1B1 tiene una actividad distinta, selectiva para la hidroxilación en posición 4 del estradiol y de la estrona (103). Este efecto inductor por beber alcohol podría aumentar las posibilidades de la participación de la vía mediada por el citocromo en relación con otra relacionada con la interacción directa de los estrógenos con ER. Es factible entonces que, en condiciones en que se producen estrés oxidativo o condiciones peroxidantes (como las reportadas por nuestro laboratorio durante la exposición de tejido mamario al alcohol), la transformación de uno a otro compuesto podría facilitarse aún más. Además, las o-quinonas son también compuestos con actividad redox potente (102)(104). Por ejemplo, estas quinonas pueden ser detoxificadas reductivamente por una quinona reductasa en un proceso de transferencia de dos electrones (105). Sin embargo, también pueden sufrir ciclos redox con la forma semiquinona de la P450 reductasa (un proceso de un electrón) y generar radicales superóxido, que en la presencia de hierro u otro metal de transición producen radicales hidroxilo (104) (Fig. 2). Por otro lado, las quinonas de estrógenos pueden dañar directamente el ADN dando lugar a efectos genotóxicos (106)(107). Los aductos de ADN de quinonas de estrógenos se han detectado en las glándulas mamarias de ratas ACI tratadas con 4-hidroxiestradiol o sus quinonas (107). Además, se han utilizado procedimientos de LC/MS-MS con alta sensibilidad recientemente desarrollados para analizar el ADN de los tumores de mama humanos y sus tejidos adyacentes y así se detectaron los aductos de ADN formados a partir de quinonas de estrógenos (108).

Figura 2. La oxidación de estrógenos a metabolitos reactivos que se unen a ADN para generar aductos depurinantes.
Todos estos resultados sugieren que estos mecanismos podrían ser relevantes para los efectos cancerígenos de los estrógenos por sí solos. Sin embargo, no hay evidencia disponible en la literatura de su formación en el tejido mamario de animales o de seres humanos expuestos al alcohol. Como parte de la presente hipótesis de trabajo acerca de la contribución de los estrógenos en los efectos del alcohol sobre el tejido mamario, se asume que se pueden formar estas quinonas de estrógenos (Fig. 2). Las razones de esa hipótesis se describirán a continuación y están relacionadas con algunos de los resultados obtenidos en este laboratorio. Para poder vincular los efectos de cooperación entre el metabolismo del alcohol en acetaldehído, su capacidad para promover el estrés oxidativo y la participación de los niveles inducidos de estrógeno en los efectos cancerígenos en el tejido mamario es necesario describir lo que sucede con los catecol-estrógenos y las estrógenoquinonas después de su formación. Los catecol-estrógenos pueden ser metabolizados por O-metilación, por reacción con glutatión, y también por glucuronidación y sufatación (109)(110).
Curiosamente, los metabolitos de catecol-estrógeno muestran una actividad estrogénica ER-mediada menos intensa, lo que implica que los catecol-estrógenos podrían tener ambos, efectos genotóxicos directos así como las acciones promotoras de tumores mediadas por ER (111). Además, los conjugados de sulfato y de glucurónido parecen jugar un papel para los estrógenos libres y, en general, existe en la actualidad una controversia sobre si el metabolismo de la conjugación puede mitigar los efectos cancerígenos mediados por catecol-estrógenos (111). No obstante, los estrógenos conjugados parecían excretarse con mayor facilidad que los estrógenos precursores lipofílicos (111). Estos sugiere que la vía de la conjugación se considera como un mecanismo de protección contra el daño causado por metabolitos reactivos de los estrógenos (109). Un aspecto importante del mecanismo de la generación de quinonas de estrógenos desde catecol-estrógenos se refiere a la formación resultante de ROS durante el proceso. En efecto, las ROS se generan a través del ciclo redox entre los catecol-estrógenos y sus análogos de quinona (112-114) (Fig. 2).
Los catecol-estrógenos pueden ser oxidados por alguna enzima o iones metálicos oxidantes tales como Cu2+ o Fe3+ para dar lugar a semiquinonas y o-quinonas (112- 114). Se plantea como hipótesis que podría muy bien ser que esta vía de activación de los catecol-estrógenos fuera estimulada durante el consumo de alcohol en el tejido mamario por el efecto inductivo del alcohol sobre la XOR y la lipoxigenasa o incluso más probablemente, porque el consumo de alcohol promueve el estrés oxidativo que se observó (17)(24)(27). La reducción de las oquinonas a semiquinonas y catecoles por la vía de la P450 reductasa proporciona una oportunidad para generar ROS, incluyendo superóxido y radicales hidroxilo (111). En los experimentos sobre el efecto de la ingesta de alcohol sobre el tejido mamario de rata pudo detectarse la formación de radicales hidroxilo e hidroperóxidos de lípidos durante el metabolismo del etanol en este tejido (18)(24). En esos estudios también se informó que el consumo de alcohol repetitivo provocó el agotamiento de las defensas contra el estrés oxidativo debido a la reducción significativa de los niveles de glutatión, de á-tocoferol y de enzimas defensivas tales como la glutatión transferasa y la glutatión reductasa (27). Además de las consecuencias que esos efectos tienen en el proceso de estrés oxidativo en sí y en la promoción del proceso carcinogénico (77), esto podría tener consecuencias adicionales relevantes para la contribución de los estrógenos en el proceso carcinogénico mamario. En efecto, el glutatión también es capaz de reaccionar con las quinonas de estrógenos y las semiquinonas para dar conjugados en una reacción catalizada por la glutatión transferasa (111). Además, los niveles bajos de glutatión también disminuyen la capacidad de tejido mamario para destruir hidroperóxidos, ya que es el cofactor necesario para la glutatión peroxidasa, para operar incluso cuando los niveles de esta enzima no se redujeron (27).
La disminución de los niveles de dos antioxidantes críticos como la vitamina E y el glutatión observada en el tejido mamario expuesto al alcohol podría ser no sólo de relevancia para la promoción de estrés oxidativo (27), sino también para comprender que su agotamiento podría ser un factor del efecto genotóxico de los estrógenos, a través de su metabolismo oxidativo que podían verse incrementado (111) (Fig. 2). El aumento de la generación de ROS producido por la toxicidad del alcohol (70) y los efectos del estrógeno (113) podrían explicar la disminución de los niveles de á-tocoferol que se observó (27). Además, la disminución en los niveles de la GST observada en los experimentos sobre el efecto de la ingesta de alcohol en el tejido mamario también puede explicarse como atribuible a los efectos inhibitorios potentes que los metabolitos de glutatión-estrógenos específicos tienen en la molécula de GST (115-117). Por supuesto esto es algo que aún no se ha demostrado. Puede ser de especial relevancia a la luz de las observaciones reportadas por Zheng et al. en los estudios epidemiológicos (118), que el consumo de alcohol incremente el riesgo de cáncer de mama en las mujeres, entre las que llevan los genotipos GST susceptibles. No sólo la vía detoxificante de los metabolitos de estrógeno mediante GSH/GST podría ser modulada negativamente por el consumo de alcohol. Otra afectada podría ser la carboximetil transferasa (COMT) y la vía de metilación de los catecol-estrógenos (Fig. 3). Se conoce ya que varios puntos del ciclo en este proceso podrían ser alterados durante el consumo de alcohol. Por ejemplo, se sabe que la S-adenosilmetionina (SAM) que participa en la etapa de la metiltransferasa que produce la metilación de los catecol-estrógenos está disminuida debido a una disminución de su biosíntesis hepática. En la enfermedad hepática alcohólica la capacidad metionina adenosiltransferasa se ve afectada. De hecho, los alcohólicos crónicos tienen hipermetioninemia y son deficientes para el clearance de metionina (119).

Figura 3. Caminos para la metilación de los catecol-estrógenos por la COMT.
Eso conduce a una disminución de la biosíntesis de SAM y a su vez impacta en la capacidad de metilación de la célula y en la disminución de las defensas antioxidantes (119). Además, se ha demostrado que la exposición crónica al etanol no sólo disminuye los niveles de hepáticas de SAM, sino también que aumenta las concentraciones hepáticas de S-adenosilhomocisteína (SAH) y disminuye las concentraciones plasmáticas de folato, en los animales y los seres humanos (119)(120). A su vez, la homocisteína puede ejercer efectos patogénicos en gran medida a través de la acumulación metabólica de SAH, que es un potente inhibidor no competitivo de los metabolismos de metilación mediados por la COMT de varios substratos de tipo catecol, incluidos los derivados de los estrógenos (121). Además del agotamiento de las concentraciones de folato, la exposición crónica de etanol disminuye la actividad de la sintasa de metionina, que se requiere para catalizar la transferencia de un grupo metilo del ácido fólico a la homocisteína para formar metionina (120)(122)(123). Todos estos hechos podrían disminuir la capacidad de la COMT para metilar catecol-estrógenos nocivos durante el consumo crónico de alcohol, como se describe en la presente hipótesis de trabajo acerca de la acción concertada de beber alcohol, la formación de acetaldehído nocivo, los radicales libres, el estrés oxidativo y el aumento de los niveles de estrógenos.
Hipótesis de trabajo sobre cómo beber alcohol puede promover efectos cancerígenos en el tejido mamario
Teniendo en cuenta la evidencia creciente sobre la relevancia de la participación del acetaldehído en los efectos carcinogénicos del alcohol en los órganos diana (por ejemplo, el tracto aero-digestivo), es razonable considerar que su formación in situ y la acumulación en el tejido mamario deben jugar un papel. La contribución del acetaldehído que llega por el torrente sanguíneo jugaría un papel secundario. Teniendo en cuenta también que se observó la promoción de estrés oxidativo y que los sistemas de generación de radicales libres están presentes en el tejido mamario expuestos al etanol durante la ingesta de alcohol, sería importante considerar el estrés oxidativo como un participante necesario en los efectos cancerígenos que el consumo de alcohol promueve en el tejido mamario. Es bien conocido que desempeña un papel en la promoción y la iniciación de tumores inducidos por otros productos químicos que actúan en otros tejidos, así como en el tejido mamario.
Por último, también se sabe que el consumo de alcohol aumenta los niveles de estrógeno en la sangre en los animales y los seres humanos y es un carcinógeno establecido para el tejido mamario (99)(124). En las mujeres, Eriksson y colaboradores observaron una elevación del acetaldehído y de estrógenos asociada después de la ingesta de alcohol (125). El alcohol es capaz también de provocar, incluso en estudios de cultivo, efectos proliferativos y de transformación de células, no sólo a través de los ER, sino también como un "carcinógeno químico", mediado por los productos de su metabolismo oxidativo (91)(126)(127). A la luz de la extraordinaria sensibilidad del tejido mamario al alcohol, claramente evidenciada en los estudios epidemiológicos, se acuñó la hipótesis de trabajo de que los efectos cancerígenos perjudiciales resultantes podrían surgir a partir de una especie de participación colaborativa de estos tres componentes, haciendo alguno de ellos más susceptibles al efecto del otro. Algunas de esas posibilidades serán descritas en el momento de proponer tratamientos preventivos potenciales, postulados sobre la base de la literatura existente vinculada. Sólo el reto de la hipótesis dirá si es válida o no. La falta de hipótesis conduce a nuevos experimentos. Nuevos experimentos pueden mostrar la necesidad de una nueva hipótesis. La presente hipótesis propuesta se muestra en la Figura 4.

Figura 4. Hipótesis de trabajo sobre el mecanismo de la promoción de cáncer de mama por la exposición a bebidas alcohólicas.
Tratamientos preventivos potenciales contra los efectos nocivos en el tejido mamario por el consumo de alcohol
El primer punto a destacar es que siendo el consumo de alcohol una de las pocas causas evitables de promoción del cáncer de mama una sabia sugerencia podría ser no beber. Sin embargo la epidemiología demuestra que el verdadero éxito de ese consejo puede no ser fácil de lograr. La actual tolerancia sugerida para beber alcohol por las mujeres es de una bebida ("trago") al día (3). El análisis de las posibles estrategias de prevención para las mujeres que deciden exceder moderadamente los niveles tolerados sugeridos se analizará a la luz de los conocimientos actuales y de la hipótesis de trabajo. Las alternativas de prevención que pueden analizarse son: 1) El bloqueo de la generación de acetaldehído y su acumulación en el tejido mamario. 2) La prevención con antioxidantes del estrés oxidativo inducido por el alcohol y por los estrógenos en el tejido mamario. 3) Los tratamientos que incrementan las defensas contra los efectos provocados por los estrógenos. 4) Los tratamientos de carácter preventivo en los diferentes aspectos del problema.
EFECTOS INHIBIDORES SOBRE LAS ENZIMAS QUE GENERAN ACETALDEHÍDO PRESENTES EN EL TEJIDO MAMARIO
Teniendo en cuenta que la mayoría del acetaldehído acumulado en el tejido mamario se generó in situ (17), es de interés particular prestar atención a los inhibidores de las dos vías metabólicas ya descriptas por este laboratorio, una presente en la fracción citosólica y mediada por XOR y otra, situada en la fracción microsomal y vinculada con una actividad de la lipoxigenasa (10)(18)(19(128-130). Es de especial interés la búsqueda de compuestos presentes en los alimentos o bebidas o disponibles como suplementos dietéticos, que tienen baja toxicidad, con efectos inhibidores potentes e idealmente que actúa sobre ambas, las vías citosólicas y las microsomales de metabolismo de etanol a acetaldehído (131)(132). Varios fitoquímicos de naturaleza polifenólica aparecieron como posibles candidatos que presentan esas características interesantes y que merecen nuestra atención en el futuro. Los inhibidores más potentes entre los polifenoles fueron los flavonoles quercetina, miricetina y kaempferol; también las flavonas apigenina y luteolina y los polifenoles ácido nordihidroguaiarético y ácido elágico. Puede ser importante en este punto tener en cuenta que se encontraron varios polifenoles que son inhibidores de la carcinogénesis en investigaciones de laboratorio (133-135). El caso del ácido elágico es de particular interés, ya que está presente en varias fuentes de frutas (136). Este compuesto fue capaz de disminuir la tumorigénesis mamaria mediada por estrógenos en ratas de la cepa ACI (137). Otros autores reportaron actividades antiproliferativas in vitro con varios tipos de células (138). Más aún, esta sustancia ha mostrado efectos beneficiosos contra el cáncer, basado en un estudio en pacientes varones con cáncer de próstata (139)(140). También se encontró que el ácido fólico se comportó como un inhibidor muy potente de la formación de acetaldehído en la fracción citosólica de tejido mamario (128). Esto sugiere un efecto preventivo mediado no sólo por el agotamiento de acetaldehído, sino también porque es un componente crítico de los procesos generales de metilación. Se encontró que el folato era relevante en la prevención de cáncer de mama (141).
LA ELIMINACIÓN DEL ACETALDEHÍDO ACUMULADO EN EL TEJIDO MAMARIO.
Como ya se ha mencionado, estudios de este laboratorio demostraron que el acetaldehído se acumula en el tejido mamario durante largos periodos de tiempo, incluso después de dosis únicas de etanol dado por vía oral (17). La eliminación del acetaldehído desde el tejido mamario aparece mucho más difícil que en el hígado. En efecto, la actividad de ALDH en las fracciones subcelulares de tejidos mamarios (citosol, mitocondrias y microsomas) fue al menos diez veces menor que en el hígado (17). Además, la actividad de ALDH mitocondrial relevante podría ser inhibida irreversiblemente por el 4HNE, subproducto de la peroxidación lipídica, que es un potente inhibidor así como un sustrato para ALDH (142)(143). La peroxidación lipídica se puede producir durante el estrés oxidativo producido por el etanol actúa sobre el tejido mamario (24). Además, el otro sistema potencialmente capaz de deshacerse de acetaldehído, GSH/GST, se reduce significativamente en el tejido mamario después de beber repetidamente alcohol (27) Otra alternativa para eliminar el acetaldehído acumulado en el tejido mamario podría surgir de la administración de cisteína. Este aminoácido es capaz de reducir la toxicidad del acetaldehído mediante la formación de un aducto estable, el ácido 2-metiltiazolidina-4- carboxílico (144). Su eficiencia para disminuir la acumulación de acetaldehído en el tejido mamario queda por establecer. Esta alternativa se emplea adecuadamente para evitar la acumulación de acetaldehído en la cavidad oral durante el consumo de alcohol (145)(146). En el caso del tejido mamario, es probable que la Nacetilcisteína (NAC) tenga una acción preventiva mayor. NAC podría ser menos eficaz que la cisteína en el medio extracelular (por ejemplo, saliva), pero se puede dar en dosis más altas y durante períodos de tiempo más largos que la cisteína, debido a su menor toxicidad y porque también es un precursor de la cisteína y el glutatión (147). En una serie de estudios desde 1984, este compuesto también ha demostrado tener el potencial para prevenir el cáncer y otras enfermedades relacionadas con mutaciones (147).
LA PREVENCIÓN DE LA OXIDACIÓN DE LOS ESTRÓGENOS O LA MEJORA DE LA DESINTOXICACIÓN DE SUS METABOLITOS REACTIVOS
La hipótesis que se muestra en la Figura 4 supone que un componente importante en la promoción de cáncer de mama por el consumo de alcohol implica de formación de catecol-estrógenos que pueden ser oxidados a sus quinonas, reactivas hacia el ADN. La contribución del consumo de alcohol a esta vía perjudicial podría implicar el efecto inductor sobre el CYP1B1 y la provisión de condiciones de oxidación relevantes para la generación de los metabolitos reactivos del estrógeno y la disminución de la desintoxicación de los metabolitos reactivos. La contribución de los antioxidantes, si esta hipótesis fuera probable, aparece atractiva para evitar que la ingesta de alcohol estimule este metabolismo oxidativo de los estrógenos. Estudios recientes mostraron que el antioxidante N-acetilcisteína, un precursor de la cisteína y del GSH intracelular es capaz de bloquear el paso inicial en la genotoxicidad causado por las estrógeno quinonas (148)(149). Los autores concluyeron que la acción preventiva de la N-acetilcisteína incluye múltiples mecanismos de protección, incluyendo nucleofilicidad, actividad antioxidante y la inhibición de la formación de los aductos de ADN (150). También se observaron propiedades preventivas en las células epiteliales de mama humanas y de ratón (151).
Además el GSH, un antioxidante ubicuo, es capaz de reaccionar no enzimáticamente con las quinonas de catecol- estrógenos o de manera más eficiente, contando con la activación catalítica de GST (150)(152). El resveratrol, un antioxidante natural presente en las uvas y en diferentes productos vegetales (153)(154) también fue eficaz en la inhibición de la formación de aductos de ADN de estrógeno (150) y también se sabe que ejerce diversos efectos anticancerígenos in vitro e in vivo (153)(155). El ácido dihidrolipoico, que se forma in vivo cuando se administra ácido lipoico, también puede inhibir la formación de aductos de ADN-estrógeno depurinantes (150). El resveratrol logró el nivel más alto de efecto inhibitorio y el NAC y el ácido dihidrolipoico una acción moderada (150). Curiosamente, este laboratorio reportó que beber alcohol disminuye los niveles de GSH, á-tocoferol y GST y mayor estrés oxidativo en el tejido mamario (27). Estos efectos deletéreos inducidos por beber alcohol deben disminuir la vía desintoxicante de la formación de conjugados de glutatión, dando a los estrógenos-3,4-quinonas más oportunidad de reaccionar con el ADN para producir aductos depurinantes. Otro efecto nocivo del consumo de alcohol también puede ocurrir a nivel de la vía desintoxicante de la COMT que opera a nivel de los catecol-estrógenos, especialmente sobre el metabolito 4-hidroxilado del 17â-estradiol. En efecto, para proceder la vía de la COMT requiere de la participación de SAM, ácido fólico, y vitaminas B6 y B12 (Fig. 3).
La metil transferasa que metila a continuación el grupo 4-OH de los catecol-estrógenos para dar el derivado 4-OCH3 requiere SAM. El producto tiene un potencial tóxico menor (102)(111). En el proceso de transmetilación mediado por SAM, la S-adenosilhomocisteína (SAH) se genera como un producto y se hidroliza posteriormente por la SAH hidrolasa para formar la homocisteína y adenosina. SAH es un potente inhibidor competitivo de la reacción de metilación, y es importante para eliminar la adenosina y homocisteína y para evitar la acumulación de SAH. La re-metilación de la homocisteína requiere ácido fólico y vitamina B12. La homocisteína también puede formar cisteína a través de procesos enzimáticos que requieren vitamina B6. La cisteína es, a su vez, el precursor limitante para la síntesis de GSH a través de un proceso de dos pasos enzimático. Todos estos procesos han sido estudiados con mucho detalle por varios autores (119)(120)(122)(156). En la Figura 3 se presenta un resumen del proceso de metilación de los catecol-estrógenos mediado por SAM. La razón para introducir brevemente este asunto es que este proceso desintoxicante por la COMT también ofrece posibilidades de tratamientos preventivos. En primer lugar, es importante resumir cuáles son los efectos del consumo de alcohol sobre los componentes de las moléculas que participan del proceso de transmetilación. Por ejemplo, se ha demostrado que la exposición crónica de etanol disminuye la concentración hepática de SAM, provoca el aumento de la concentración plasmática de homocisteína, aumenta la concentración hepática de SAH y disminuye la concentración plasmática de folato, en estudios en humanos y animales (120).
Por el contrario, la administración de SAM se ha utilizado para atenuar los efectos nocivos del consumo de alcohol en los animales. La administración SAM es conocida por restaurar las concentraciones hepáticas de GSH mermadas por el consumo de alcohol (120) Es decir, el tratamiento SAM también puede ser de ayuda para prevenir los efectos de beber alcohol en el cáncer de mama inducido por la mejora de la vía de desintoxicación por la COMT.
Consideraciones finales
Las consideraciones sobre la patogénesis del cáncer mamario inducido por beber alcohol llevaron a la hipótesis de trabajo de que: la acumulación de acetaldehído y sus efectos nocivos, la generación de ROS inducida y efectos deletéreos oxidativos sobre el aumento de los niveles de estrógeno en cooperación podrían estar involucrados en la promoción del cáncer. Eso está por demostrarse. Si las suposiciones fueran aún correctas parcialmente, algunas alternativas preventivas útiles podrían aprovecharse: la inhibición de las vías metabólicas que generan acetaldehído, la destrucción del acetaldehído acumulado, los tratamientos con antioxidantes potentes y seguros y también mejorar las vías desintoxicantes de quinonas de estrógenos. Además, se podrían contemplar cócteles que tienen mezclas de componentes de cada tipo.
No obstante, siempre es importante tener en cuenta que el cáncer de mama inducido por el consumo de alcohol es una de las pocas causas evitables de cáncer y que sería posible evitar beber alcohol en absoluto, o más probablemente, beber sólo una copa al día (12 gramos de etanol por día solamente).
1. World Health Organization. Global Status Report on Alcohol and Health. Geneva: WHO Press; 2014. [ Links ]
2. International Agency for Research on Cancer. Alcohol Drinking. IARC Monogr Eval Carcinog Risks Hum 1988; 44: 7-378. [ Links ]
3. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Alcohol consumption and ethyl carbamate. IARC Monogr Eval Carcinog Risks Hum 2010; 96: 3-1383. [ Links ]
4. Allen NE, Beral V, Casabonne D, Kan SW, Reeves GK, Brown A, et al. Million Women Study Collaborators. Moderate alcohol intake and cancer incidence in women. J Natl Cancer Inst 2009; 101: 296-305. [ Links ]
5. Hamajima N, Hirose K, Tajima K, Rohan T, Calle EE, Heath CW Jr, et al. Collaborative Group on Hormonal Factors in Breast Cancer. Alcohol, tobacco and breast cancer-collaborative reanalysis of individual data from 53 epidemiological studies, including 58, 515 women with breast cancer and 95,067 women without the disease. Br J Cancer 2002; 87: 1234-45. [ Links ]
6. Seitz HK, Pelucchi C, Bagnardi V, La Vecchia C. Epidemiology and pathophysiology of alcohol and breast cancer: update 2012. Alcohol Alcohol 2012; 47: 204-12. [ Links ]
7. Schütze M, Boeing H, Pischon T, Rehm J, Kehoe T, Gmel G, et al. Alcohol attributable burden of incidence of cancer in eight European countries based on results from prospective cohort study. BMJ 2011; 342: d1584. [ Links ]
8. Stewart BW, Kleihues P. WHO - International Agency for Research on Cancer. World Cancer Report. Lyon: IARC Press; 2003.p. 29-32. [ Links ]
9. Dumitrescu RG, Shields PG. The etiology of alcohol-induced breast cancer. Alcohol 2005; 35: 213-25. [ Links ]
10. Castro GD, Castro JA. Metabolism of ethanol to acetaldehyde in the rat mammary tissue. Inhibitory effects of plant polyphenols and folic acid. En: Watson RR, Preedy VR, Zibadi S, editores. Alcohol, Nutrition and Health Consequences. Nutrition and Health. New York: Springer Science + Business Media; 2013. p. 145-54. [ Links ]
11. Castro GD, Quintans LN, Maciel ME, Castro JA. Preventive effects of plant polyphenols in the promotion of mammary cancer and testicular damage induced by alcohol drinking. En: Watson RR, Preedy VR, Zibadi S, editores. Polyphenols in Human Health and Disease. San Diego: Elsevier-Academic Press; 2014. p. 1181-90. [ Links ]
12. Garro AJ, Lieber CS. Alcohol and cancer. Annu Rev Pharmacol Toxicol 1990; 30: 219-49. [ Links ]
13. Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr 2010; 5: 121-8. [ Links ]
14. Dellarco VL. 1988. A mutagenicity assessment of acetaldehyde. Mutat Res 1988; 195: 1-20. [ Links ]
15. Woutersen RA, Appelman LM, Feron VJ, Van der Heijden CA. Inhalation toxicity of acetaldehyde in rats. II. Carcinogenicity study: interim results after 15 months. Toxicology 1984; 31: 123-33. [ Links ]
16. Woutersen RA, Appelman LM, Van Garderen-Hoetmer A, Feron VJ. Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology 1986; 41: 213-31. [ Links ]
17. Castro GD, Delgado de Layño AM, Fanelli SL, Maciel ME, Díaz Gómez MI, Castro JA. Acetaldehyde accumulation in rat mammary tissue after an acute treatment with alcohol. J Appl Toxicol 2008; 28: 315-21. [ Links ]
18. Castro GD, Delgado de Layño AMA, Costantini MH, Castro JA. Cytosolic xanthine oxidoreductase mediated bioactivation of ethanol to acetaldehyde and free radicals in rat breast tissue. Its potential role in alcohol promoted mammary cancer. Toxicology 2001; 160: 11-8. [ Links ]
19. Castro GD, Delgado de Layño AMA, Costantini MH, Castro JA. Rat breast microsomal biotransformation of ethanol to acetaldehyde but not to free radicals. Its potential role in the association between alcohol drinking and breast tumor promotion. Teratog Carcinog Mutagen 2003; 23 Suppl 1: 61-70. [ Links ]
20. Fam AG. Gout: excess calories, purines, and alcohol intake and beyond. Response to a urate-lowering diet. J Rheumatol 2005; 32: 773-7. [ Links ]
21. Lieber CS. Alcohol metabolism: general aspects. En: Watson RR, Preedy VR, editores. Comprehensive Handbook of Alcohol Related Pathology, Volume 1. London: Elsevier Science Ltd-Academic Press; 2005. p. 15-26. [ Links ]
22. Jarasch ED, Grund C, Bruder G, Heid HW, Keenan TW, Franke WW. Localization of xanthine oxidase in mammary gland epithelium and capillary endothelium. Cell 1981; 25: 67-82. [ Links ]
23. Kooij A, Frederiks WM, Gossrau R, Van Noorden CJ. Localization of xanthine oxidoreductase activity using the tissue protectant polyvinyl alcohol and final electron acceptor Tetranitro BT. J Histochem Cytochem 1991; 39: 87-93. [ Links ]
24. Castro GD, de Castro CR, Maciel ME, Fanelli SL, de Ferreyra EC, Díaz Gómez MI, et al. Ethanol-induced oxidative stress and acetaldehyde formation in rat mammary tissue: Potential factors involved in alcohol drinking promotion of breast cancer. Toxicology 2006; 219: 208-19. [ Links ]
25. Guerri C, Sanchis R. Alcohol and acetaldehyde in rat's milk following ethanol administration. Life Sci 1986; 28: 1543-56. [ Links ]
26. Triano EA, Slusher LB, Atkins TA, Beneski JT, Gestl SA, Zolfaghari R, et al. Class I alcohol dehydrogenase is highly expressed in normal human mammary epithelium but not in invasive breast cancer: implications for breast carcinogenesis. Cancer Res 2003; 63: 3092-100. [ Links ]
27. Fanelli SL, Maciel ME, Díaz Gómez MI, Delgado de Layño AM, Bietto FM, Castro JA, et al. Further studies in the potential contribution of acetaldehyde accumulation and oxidative stress in rat mammary tissue in the alcohol drinking promotion of breast cancer. J Appl Toxicol 2011; 31: 11-9. [ Links ]
28. O'Brien PJ. Peroxidases. Chem Biol Interact 2000; 129: 113-39. [ Links ]
29. Natarajan R, Nadler J. Role of lipoxygenases in breast cancer. Front Biosci 1998; 3: E81-8. [ Links ]
30. Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry 1999; 38: 3694-703. [ Links ]
31. Fang JL, Vaca CE. Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis 1997; 18: 627-32. [ Links ]
32. Díaz Gómez MI, Fanelli SL, Castro GD, Costantini MH, Castro JA. A liver nuclear ethanol metabolizing system. Formation of metabolites that bind covalently to macromolecules and lipids. Toxicology 1999; 138: 19-28. [ Links ]
33. Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Identification of DNA adducts of acetaldehyde. Chem Res Toxicol 2000; 13: 1149-57. [ Links ]
34. Balbo S, Hashibe M, Gundy S, Brennan P, Canova C, Simonato L, et al. N2-ethyldeoxyguanosine as a potential biomarker for assessing effects of alcohol consumption on DNA Cancer. Epidemiol Biomarkers Prev 2008; 17: 3026-32. [ Links ]
35. Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M, et al. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact 2010; 188: 367-75. [ Links ]
36. Matsuda T, Yabushita H, Kanaly RA, Shibutani S, Yokoyama A. Increased DNA damage in ALDH2-deficient alcoholics. Chem Res Toxicol 2006; 19: 1374-8. [ Links ]
37. Lorenti Garcia C, Mechilli M, Proietti De Santis L, Schinoppi A, Kobos K, Palitti F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat Res 2009; 662: 3-9. [ Links ]
38. Freudenheim JL, Bonner M, Krishnan S, Ambrosone CB, Graham S, McCann SE, et al. Diet and alcohol consumption in relation to p53 mutations in breast tumors. Carcinogenesis 2004; 25: 931-9. [ Links ]
39. Niemelä O. Distribution of ethanol-induced protein adducts in vivo: relationship to tissue injury. Free Radic Biol Med 2001; 31: 1533-8. [ Links ]
40. Castro GD, Delgado de Layño AM, Castro JA. Liver nuclear ethanol metabolizing systems (NEMS) producing acetaldehyde and 1-hydroxyethyl free radicals. Toxicology 1998; 129: 137-44. [ Links ]
41. Castro GD, Díaz Gómez MI, Castro JA. Species differences in the interaction between CCl4 reactive metabolites and liver DNA or nuclear protein fractions. Carcinogenesis 1989; 10: 289-94. [ Links ]
42. Díaz Gómez MI, Valles E, Fanelli SL, de Layño AM, Castro GD, Castro JA. Alcohol induction of liver nuclear ethanol and N-nitrosodimethylamine metabolism to reactive metabolites. Teratog Carcinog Mutagen 2002; 22: 139-45. [ Links ]
43. Díaz Gómez MI, Fanelli SL, Delgado de Layño AM, Bietto FM, Castro JA, Castro GD. Deleterious effects induced by oxidative stress in liver nuclei from rats receiving an alcohol-containing liquid diet. Toxicol Ind Health 2008; 24: 625-34. [ Links ]
44. Dasso M. The role of the Ran GTPase pathway in cell cycle control and interphase nuclear functions. Prog Cell Cycle Res 1995; 1: 163-72. [ Links ]
45. He D, Zeng C, Brinkley BR. Nuclear matrix proteins as structural and functional components of the mitotic apparatus. Int Rev Cytol 1995; 162B: 1-74. [ Links ]
46. Martelli AM, Bareggi R, Bortul R, Grill V, Narducci P, Zweyer M. The nuclear matrix and apoptosis. Histochem Cell Biol 1997; 108: 1-10. [ Links ]
47. Oliff A, Gibbs JB, McCormick F. New molecular targets for cancer therapy. Sci Am 1996; 275: 144-9. [ Links ]
48. Weinberg RA. How cancer arises. Sci Am 1996; 275: 62-70. [ Links ]
49. Garro AJ, Espina N, Farinati F, Salvagnini M. The effects of chronic ethanol consumption on carcinogen metabolism and on O6-methylguanine transferase-mediated repair of alkylated DNA. Alcohol Clin Exp Res 1986; 10: 73S-7S. [ Links ]
50. Espina N, Lima V, Lieber CS, Garro AJ. In vitro and in vivo inhibitory effect of ethanol and acetaldehyde on O6-methylguanine transferase. Carcinogenesis 1988; 9: 761-6. [ Links ]
51. Mufti SI, Salvagnini M, Lieber CS, Garro AJ. Chronic ethanol consumption inhibits repair of dimethylnitrosamine- induced DNA alkylation. Biochem Biophys Res Commun 1988; 152: 423-31. [ Links ]
52. Mikami K, Haseba T, Ohno Y. Ethanol induces transient arrest of cell division (G2 + M block) followed by G0/G1 block: dose effects of short- and longer-term ethanol exposure on cell cycle and cell functions. Alcohol Alcohol 1997; 32: 145-52. [ Links ]
53. Kenney WC. Acetaldehyde adducts of phospholipids. Alcohol Clin Exp Res 1982; 6: 412-6. [ Links ]
54. Kenney WC. Formation of Schiff base adduct between acetaldehyde and rat liver microsomal phosphatidylethanolamine. Alcohol Clin Exp Res 1984; 8: 551-5. [ Links ]
55. Fanelli SL, Castro GD, Castro JA. Cholesterol interaction with free radicals produced from carbon tetrachloride or bromotrichloromethane by either catalytic decomposition or via liver microsomal activation. Chem Biol Interact 1995; 98: 223-36. [ Links ]
56. Fanelli SL, Castro JA. Covalent binding of carbon tetrachloride reactive metabolites to liver microsomal and nuclear lipid and phospholipid clases from Sprague Dawley and Osborne Mendel male rats. Teratog Carcinog Mutagen 1995; 15: 155-66. [ Links ]
57. Goodman DS, Deykin D. Fatty acid ethyl ester formation during ethanol metabolism in vivo. Proc Soc Exp Biol Med 1963; 113: 65-7. [ Links ]
58. Lange LG. Nonoxidative ethanol metabolism: formation of fatty acid ethyl esters by cholesterol esterase. Proc Natl Acad Sci USA 1982; 79: 3954-7. [ Links ]
59. Laposata M. Fatty acid ethyl esters: ethanol metabolites which mediate ethanol-induced organ damage and serve as markers of ethanol intake. Prog Lipid Res 1998; 37: 307-16. [ Links ]
60. Sun GY, Sun AY. Ethanol and membrane lipids. Alcohol Clin Exp Res 1985; 9: 164-80. [ Links ]
61. Sylvia VL, Joe CO, Norman JO, Curtin GM, Busbee DL. Phosphatidylinositol-dependent activation of DNA polymerase alpha. Biochem Biophys Res Commun 1986; 135: 880-5. [ Links ]
62. Herlan G, Giese G, Wunderlich F. Influence of nuclear membrane lipid fluidity on nuclear RNA release. Exp Cell Res 1979; 118: 305-9. [ Links ]
63. Cocco L, Gilmour RS, Ognibene A, Letcher AJ, Manzoli FA, Irvine RF. Synthesis of polyphosphoinositides in nuclei of Friend cells. Evidence for polyphosphoinositide metabolism inside the nucleus which changes with cell differentiation. Biochem J 1987; 248: 765-70. [ Links ]
64. Venkatraman JT, Lefebvre YA, Clandinin MT. Diet fat alters the structure and function of the nuclear envelope: modulation of membrane fatty acid composition, NTPase activity and binding of triiodothyronine. Biochem Biophys Res Commun 1986; 135: 655-61. [ Links ]
65. Venkatraman JT, Clandinin MT. Ribonucleic acid efflux from isolated mouse liver nuclei is altered by diet and genotypically determined change in nuclear envelope composition. Biochim Biophys Acta 1988; 940: 33-42. [ Links ]
66. Manzoli FA, Martelli AM, Capitani S, Maraldi NM, Rizzoli R, Barnabei O, et al. Nuclear polyphosphoinositides during cell growth and differentiation. Adv Enzyme Regul 1989; 28: 25-34. [ Links ]
67. Chapkin RS, Davidson LD, Davidson LA. Phospholipid molecular species composition of mouse liver nuclei. Influence of dietary n-3 fatty acid ethyl esters. Biochem J 1992; 287: 237-40. [ Links ]
68. Bartel LC, Montalto de Mecca M, Castro JA. Nitroreductive metabolic activation of some carcinogenic nitro heterocyclic food contaminants in rat mammary tissue cellular fractions. Food Chem Toxicol 2009; 47: 140-4. [ Links ]
69. Bartel LC, Montalto de Mecca M, de Castro CR, Bietto FM, Castro JA. Metabolization of nifurtimox and benznidazole in cellular fractions of rat mammary tissue. Hum Exp Toxicol 2010; 29: 813-22. [ Links ]
70. Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 2009; 83: 519-48. [ Links ]
71. Boyer PD. Sulfhydryl and Disulfide Groups of Enzymes. En: Boyer PD, Lardy H, Myrbäck K, editores. The Enzymes, 2nd ed, vol 1. New York: Academic Press; 1959. p. 512-88. [ Links ]
72. Johnstone RM. Sulfhydryl Agents: Arsenicals. En: Hochster RM, Quastel JH, editores. Metabolic Inhibitors, volume II. London: Academic Press; 1963. p. 99- 118. [ Links ]
73. Peters R. Inhibitors of enzymes containing thiol groups and toxicity. En: Biochemical Lesions and Lethal Synthesis. Volume 18, New York: Pergamon Press; 1963. p. 74-87. [ Links ]
74. Dixon M, Webb EC. Enzymes. New York: Academic Press; 1979. p. 301-7. [ Links ]
75. Malins DC, Polissar NL, Gunselman SJ. Progression of human breast cancers to the metastatic state is linked to hydroxyl radical-induced DNA damage. Proc Natl Acad Sci U S A 1996; 93: 2557-63. [ Links ]
76. Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol 2004; 44: 239-67. [ Links ]
77. Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J 2007; 401: 1-11. [ Links ]
78. Klaunig JE, Wang Z, Pu X, Zhou S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol 2011; 254: 86-99. [ Links ]
79. Reichman ME, Judd JT, Longcope C, Schatzkin A, Clevidence BA, Nair PP, et al. Effects of alcohol consumption on plasma and urinary hormone concentrations in premenopausal women. J Natl Cancer Inst 1993; 85: 722-7. [ Links ]
80. Ginsburg ES, Mello NK, Mendelson JH, Barbieri RL, Teoh SK, Rothman M, et al. Effects of alcohol ingestion on estrogens in postmenopausal women. JAMA 1996; 276: 1747-51. [ Links ]
81. Purohit V. Moderate alcohol consumption and estrogen levels in postmenopausal women: a review. Alcohol Clin Exp Res 1998; 22: 994-7. [ Links ]
82. Sarkola T, Mäkisalo H, Fukunaga T, Eriksson CJ. Acute effect of alcohol on estradiol, estrone, progesterone, prolactin, cortisol, and luteinizing hormone in premenopausal women. Alcohol Clin Exp Res 1999; 23: 976-82. [ Links ]
83. Sarkola T, Fukunaga T, Mäkisalo H, Peter Eriksson CJ. Acute effect of alcohol on androgens in premenopausal women. Alcohol Alcohol 2000; 35: 84-90. [ Links ]
84. Sarkola T, Adlercreutz H, Heinonen S, von Der Pahlen B, Eriksson CJ. The role of the liver in the acute effect of alcohol on androgens in women. J Clin Endocrinol Metab 2001; 86: 1981-5. [ Links ]
85. Coutelle C, HoHn B, Benesova M, Oneta CM, Quattrochi P, Roth HJ, et al. Risk factors in alcohol associated breast cancer: alcohol dehydrogenase polymorphysm and estrogens. Int J Oncol 2004; 25: 1127-32. [ Links ]
86. Mahabir S, Baer DJ, Johnson LL, Dorgan JF, Campbell W, Brown E, et al. The effects of moderate alcohol supplementation on estrone sulfate and DHEAS in postmenopausal women in a controlled feeding study. Nutr J 2004; 3: 11. [ Links ]
87. Sierksma A, Sarkola T, Eriksson CJ, van der Gaag MS, Grobbee DE, Hendriks HF. Effect of moderate alcohol consumption on plasma dehydroepiandrosterone sulfate, testosterone, and estradiol levels in middle-aged men and postmenopausal women: a diet-controlled intervention study. Alcohol Clin Exp Res 2004; 28: 780-5. [ Links ]
88. Seitz HK, Maurer B. The relationship between alcohol metabolism, estrogen levels, and breast cancer risk. Alcohol Res Health 2007; 30: 42-3. [ Links ]
89. Singletary KW, Gapstur SM. Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms. JAMA 2001; 286: 2143-51. [ Links ]
90. Gavaler JS, Van Thiel DH. Hormonal status of postmenopausal women with alcohol-induced cirrhosis: further findings and a review of the literature. Hepatology 1992; 16: 312-9. [ Links ]
91. Etique N, Flament S, Lecomte J, Grillier-Vuissoz I. Ethanol- induced ligand-independent activation of ERalpha mediated by cyclic AMP/PKA signaling pathway: an in vitro study on MCF-7 breast cancer cells. Int J Oncol 2007; 31: 1509-18. [ Links ]
92. Sarkar DK, Liehr JG, Singletary KW. Role of estrogen in alcohol promotion of breast cancer and prolactinomas. Alcohol Clin Exp Res 2001; 25: 230S-6S. [ Links ]
93. Roy D, Liehr JG. Estrogen, DNA damage and mutations. Mutat Res 1999; 424: 107-15. [ Links ]
94. Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents-DNA adducts and mutations. J Natl Cancer Inst Monogr 2000; 27: 75-93. [ Links ]
95. Cavalieri EL, Rogan EG. A unifying mechanism in the initiation of cancer and other diseases by catechol quinones Ann N Y Acad Sci 2004; 1028: 247-57. [ Links ]
96. Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol 2011; 125: 169-80. [ Links ]
97. Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med 2006; 354: 270-82. [ Links ]
98. Fernández SV. Estrogen, alcohol consumption, and breast cancer. Alcohol Clin Exp Res 2011; 35: 389-91. [ Links ]
99. Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis 1996; 17: 2279-84. [ Links ]
100. Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis 2000; 21: 427-33. [ Links ]
101. Flötotto T, Djahansouzi S, Gläser M, Hanstein B, Niederacher D, Brumm C, et al. Hormones and hormone antagonists: mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm Metab Res 2001; 33: 451-7. [ Links ]
102. Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol 2008; 21: 93-101. [ Links ]
103. Zhu BT, Lee AJ. NADPH-dependent metabolism of 17â-estradiol and estrone to polar and nonpolar metabolites by human tissues and cytochrome P450 isoforms. Steroids 2005; 70: 225-44. [ Links ]
104. Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol 2000; 13: 135-60. [ Links ]
105. Gaikwad NW, Yang L, Rogan EG, Cavalieri EL. Evidence for NQO2-mediated reduction of the carcinogenic estrogen ortho-quinones. Free Radic Biol Med 2009; 46: 253-62. [ Links ]
106. Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta 2006; 1766: 63-78. [ Links ]
107. Li KM, Todorovic R, Devanesan P, Higginbotham S, Köfeler H, Ramanathan R, et al. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis 2004; 25: 289-97. [ Links ]
108. Embrechts J, Lemière F, Van Dongen W, Esmans EL, Buytaert P, Van Marck E, et al. Detection of estrogen DNA-adducts in human breast tumor tissue and healthy tissue by combined nano LC-nano ES tandem mass spectrometry. J Am Soc Mass Spectrom 2003; 14: 482-91. [ Links ]
109. Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr 2000; 27: 113-24. [ Links ]
110. Hachey DL, Dawling S, Roodi N, Parl FF. Sequential action of phase I and II enzymes cytochrome p450 1B1 and glutathione S-transferase P1 in mammary estrogen metabolism. Cancer Res 2003; 63: 8492-9. [ Links ]
111. Chang M. Dual roles of estrogen metabolism in mammary carcinogenesis. BMB Rep 2011; 44: 423-34. [ Links ]
112. Zhang F, Bolton JL. Synthesis of the equine estrogen metabolites 2-hydroxyequilin and 2-hydroxyequilenin. Chem Res Toxicol 1999; 12: 200-3. [ Links ]
113. Roy D, Bernhardt A, Strobel HW, Liehr JG. Catalysis of the oxidation of steroid and stilbene estrogens to estrogen quinone metabolites by the beta-naphthoflavone- inducible cytochrome P450 IA family. Arch Biochem Biophys 1992; 296: 450-6. [ Links ]
114. Markides CS, Roy D, Liehr JG. Concentration dependence of prooxidant and antioxidant properties of catecholestrogens. Arch Biochem Biophys 1998; 360:105-12. [ Links ]
115. Abel EL, Lyon RP, Bammler TK, Verlinde CL, Lau SS, Monks TJ, et al. Estradiol metabolites as isoform-specific inhibitors of human glutathione S-transferases. Chem Biol Interact 2004; 151: 21-32. [ Links ]
116. Yao J, Chang M, Li Y, Pisha E, Liu X, Yao D, et al. Inhibition of cellular enzymes by equine catechol estrogens in human breast cancer cells: specificity for glutathione S-transferase P1-1. Chem Res Toxicol 2002; 15: 935-42. [ Links ]
117. Chang M, Shin YG, van Breemen RB, Blond SY, Bolton JL. Structural and functional consequences of inactivation of human glutathione S-transferase P1-1 mediated by the catechol metabolite of equine estrogens, 4-hydroxyequilenin. Biochemistry 2001; 40: 4811-20. [ Links ]
118. Zheng T, Holford TR, Zahm SH, Owens PH, Boyle P, Zhang Y, et al. Glutathione S-transferase M1 and T1 genetic polymorphisms, alcohol consumption and breast cancer risk. Br J Cancer 2003; 88: 58-62. [ Links ]
119. Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res 2008; 32: 1525-34. [ Links ]
120. Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am J Clin Nutr 2007; 86: 14-24. [ Links ]
121. Zhu BT. On the mechanism of homocysteine pathophysiology and pathogenesis: a unifying hypothesis. Histol Histopathol 2002; 17: 1283-91. [ Links ]
122. Kharbanda KK. Alcoholic liver disease and methionine metabolism. Semin Liver Dis 2009; 29: 155-65. [ Links ]
123. Zakhari S. Alcohol metabolism and epigenetic changes. Alcohol Res 2013; 35: 6-16. [ Links ]
124. Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J Natl Cancer Inst 1998; 90: 814-23. [ Links ]
125. Eriksson CJ, Fukunaga T, Sarkola T, Lindholm H, Ahola L. Estrogen-related acetaldehyde elevation in women during alcohol intoxication. Alcohol Clin Exp Res 1996; 20: 1192-5. [ Links ]
126. Etique N, Grillier-Vuissoz I, Lecomte J, Flament S. Crosstalk between adenosine receptor (A2A isoform) and ERalpha mediates ethanol action in MCF-7 breast cancer cells. Oncol Rep 2009; 21: 977-81. [ Links ]
127. Izevbigie EB, Ekunwe SI, Jordan J, Howard CB. 2002. Ethanol modulates the growth of human breast cancer cells in vitro. Exp Biol Med 2002; 227: 260-5. [ Links ]
128. Maciel ME, Castro GD, Castro JA. Inhibition of the rat breast cytosolic bioactivation of ethanol to acetaldehyde by some plant polyphenols and folic acid. Nutr Cancer 2004; 49: 94-9. [ Links ]
129. Maciel ME, Castro JA, Castro GD. Inhibition of rat mammary microsomal oxidation of ethanol to acetaldehyde by plant polyphenols. Hum Exp Toxicol 2011; 30: 656-64. [ Links ]
130. Hong J, Smith TJ, Ho CT, August DA, Yang CS. Effects of purified green and black tea polyphenols on cyclooxygenase- and lipoxygenase-dependent metabolism of arachidonic acid in human colon mucosa and colon tumor tissues. Biochem Pharmacol 2001; 62: 1175-83. [ Links ]
131. Cai Y, Luo Q, Sun M, Corke H. Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sci 2004; 74: 2157-84. [ Links ]
132. Cos P, Ying L, Calomme M, Hu JP, Cimanga K, Van Poel B, et al. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J Nat Prod 1998; 61: 71-6. [ Links ]
133. Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr 2001; 21: 381-406. [ Links ]
134. Lambert JD, Hong J, Yang GY, Liao J, Yang CS. Inhibition of carcinogenesis by polyphenols: evidence from laboratory investigations. Am J Clin Nutr 2005; 81: 284S-91S. [ Links ]
135. Collins-Burow BM, Burow ME, Duong BN, McLachlan JA. Estrogenic and antiestrogenic activities of flavonoid phytochemicals through estrogen receptor binding-dependent and -independent mechanisms. Nutr Cancer 2000; 38: 229-44. [ Links ]
136. Valentovic M, Santanam N. Nutrition, oxidative stress and cancer. En: Claudio PP, Niles RM, editores. Nutrition and Cancer from Epidemiology to Biology. Bentham E Books; 2012. p. 77-86. [ Links ]
137. Aiyer HS, Srinivasan C, Gupta RC. Dietary berries and ellagic acid diminish estrogen-mediated mammary tumorigenesis in ACI rats. Nutr Cancer 2008; 60: 227-34. [ Links ]
138. Losso JN, Bansode RR, Trappey A 2nd, Bawadi HA, Truax R. In vitro anti-proliferative activities of ellagic acid. J Nutr Biochem 2004; 15: 672-8. [ Links ]
139. Bell C, Hawthorne S. Ellagic acid, pomegranate and prostate cancer - A mini review. J Pharm Pharmacol 2008; 60: 139-44. [ Links ]
140. Pantuck AJ, Leppert JT, Zomorodian N, Aronson W, Hong J, Barnard RJ, et al. Phase II study of pomegranate juice for men with rising prostate-specific antigen following surgery or radiation for prostate cancer. Clin Cancer Res 2006; 12: 4018-26. [ Links ]
141. Larsson SC, Giovannucci E, Wolk A. Folate and risk of breast cancer: a meta-analysis. J Natl Cancer Inst 2007; 99: 64-76. [ Links ]
142. Deitrich RA, Petersen D, Vasiliou V. Removal of acetaldehyde from the body. En: Novartis Foundation Symposium 285. Acetaldehyde-related pathology: bridging the trans-disciplinary divide. Chichester: Wiley; 2007. p. 23-51. [ Links ]
143. Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon- 2-enal and 4-oxonon-2-enal. Chem Res Toxicol 2006; 19: 102-10. [ Links ]
144. Cederbaum AI, Rubin E. Protective effect of cysteine on the inhibition of mitochondrial functions by acetaldehyde. Biochem Pharmacol 1976; 25: 963-73. [ Links ]
145. Salaspuro VJ, Hietala JM, Marvola ML, Salaspuro MP. Eliminating carcinogenic acetaldehyde by cysteine from saliva during smoking. Cancer Epidemiol Biomarkers Prev 2006; 15: 146-9. [ Links ]
146. Salaspuro V. Pharmacological treatments and strategies for reducing oral and intestinal acetaldehyde. En: Novartis Foundation Symposium 285. Acetaldehyde- related pathology: bridging the trans-disciplinary divide. Chichester: Wiley; 2007. p. 145-57. [ Links ]
147. De Flora S, Izzotti A, D'Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis 2001; 22: 999-1013. [ Links ]
148. Zahid M, Saeed M, Ali MF, Rogan EG, Cavalieri EL. N-acetylcysteine blocks formation of cancer-initiating estrogen-DNA adducts in cells. Free Radic Biol Med 2010; 49: 392-400. [ Links ]
149. Reed DJ. Glutathione: toxicological implications. Annu Rev Pharmacol Toxicol 1990; 30: 603-31. [ Links ]
150. Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem Res Toxicol 2007; 20: 1947-53. [ Links ]
151. Venugopal D, Zahid M, Mailander PC, Meza JL, Rogan EG, Cavalieri EL, et al. Reduction of estrogen-induced transformation of mouse mammary epithelial cells by N-acetylcysteine. J Steroid Biochem Mol Biol 2008; 109: 22-30. [ Links ]
152. Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho- quinones. Free Radic Biol Med 2007; 43: 1289-98. [ Links ]
153. Aziz MH, Kumar R, Ahmad N. Cancer chemoprevention by resveratrol: in vitro and in vivo studies and the underlying mechanisms. Int J Oncol 2003; 23: 17-28. [ Links ]
154. Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997; 275: 218-20. [ Links ]
155. Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer 2003; 3: 768-80. [ Links ]
156. Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007; 45: 1306-12. [ Links ]
Recibido: 13 de mayo de 2014
Aceptado: 19 de septiembre de 2014
