










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.50 no.1 La Plata mar. 2016
TOXICOLOGÍA
Detección del alucinógeno 25C-NBOMe en orina por dos técnicas cromatográficas
Detection of hallucinogen 25C-NBOMe in urine by two chromatographic techniques
Detecção de alucinógeno 25C-NBOME na urina através de duas técnicas cromatográficas
Boris Ettienne Duffau Garrido1, Cristián Camargo Grandón2, Lorena Delgado Rivera3
1 Químico Farmacéutico. Instituto de Salud Pública de Chile, Avenida Marathon 1000 Ñuñoa Santiago de Chile.
2 Químico farmacéutico. Doctorado Facultad de Ciencias Químicas y Farmacéuticas. Universidad de Chile.
3 Q.F. Magister. Sección Química de Alimentos Instituto de Salud Pública de Chile.
CORRESPONDENCIA BORIS ETTIENNE DUFFAU GARRIDO Instituto de Salud Pública de Chile, Avenida Marathon 1000 Ñuñoa, SANTIAGO DE CHILE, Chile E-mail: bduffau@ispch.cl
Resumen
El consumo de drogas de síntesis ha ido en aumento. Estos nuevos derivados sintéticos son análogos estructurales de la feniletilamina N-sustituida. Este grupo ha provocado severos casos de intoxicación e incluso probablemente la muerte de varios consumidores. El principal derivado es conocido como 25C-NBOMe y se consume en estampillas idénticas al LSD. En este trabajo se desarrolla una metodología analítica para la determinación de 25C-NBOMe mediante cromatografía planar instrumental (cromatografía en capa delgada de alta resolución) y cromatografía de gases con detector de masas (CG/EM) como técnicas alternativas de fácil manejo y costo. Estas metodologías demostraron ser robustas y confiables para el propósito previsto.
Palabras clave: Alucinógenos; 25C-NBOMe; Extracción en fase sólida; Cromatografía en capa delgada de alta resolución; Cromatografía de gases con detector de masas.
Summary
Consumption of synthetic drugs has increased. These new synthetic derivatives are structural analogs of N-substituted phenylethylamine, and this group has caused severe cases of poisoning and even probably the death of several users. The main derivative is known as 25C-NBOMe and it is consumed in blotters in the same manner as LSD. In this work, an analytical methodology for 25C-NBOMe determination by instrumental planar chromatography high-performance thin-layer chromatography (HPTLC) and gas chromatography with mass detector (GC/MS) were developed as alternative techniques; they are easy to use and low cost. These methods proved to be robust and reliable for the intended purpose.
Key words: Allucinogens; 25C-NBOMe; Solid phase extraction (SPE); High-performance thin-layer chromatography (HPTLC); Gas chromatography with mass detector.
Resumo
O consumo de drogas sintéticas vem aumentando. Esses novos derivados sintéticos são análogos estruturais de feniletilamina N-substituída, este grupo tem causado casos graves de intoxicação e, até mesmo, provavelmente, a morte de vários consumidores. O principal derivado é conhecido como 25C-NBOMe e consumido em selos idênticos ao LSD. Neste trabalho é desenvolvida uma metodologia analítica para a determinação de 25C-NBOMe através de cromatografia planar instrumental (cromatografia em camada delgada de alta resolução) e cromatografia gasosa com detector de massas (CG/EM) como técnicas alternativas de fácil utilização e custo. Estas metodologias demonstraram serem robustas e fiáveis para a finalidade a que se destinam.
Palavras-chave: Alucinógenos; 25C-NBOMe; Extração em fase sólida; Cromatografia em camada delgada de alta resolução; Cromatografía gasosa com detector de massas.
Introducción
Desde hace un tiempo el consumo de las drogas de síntesis ha ido en aumento (1); estos derivados sintéticos son análogos estructurales de feniletilaminas N-sustituidas con el grupo 2-metoxibencilo y presentan propiedades estimulantes y alucinógenas, tienen una gran afinidad por el receptor 5HT2A, inclusive mayor que la dietilamida del ácido lisérgico (LSD), pero además poseen afinidad por los receptores α1 adrenérgicos y D1-3 de dopamina lo que explicaría su marcado efecto estimulante (2). Estas nuevas drogas se presentan en estampillas y contienen principalmente como componente activo conocido a 2-(4-cloro-2,5-dimetoxifenil)-N-[(2-metoxifenil) metil] etanamina, también denominado 25C-NBOMe, (C18H22ClNO3, peso molecular 335 g/mol).
Las dosis administradas son muy variables y no están estandarizadas (3), siendo la vía de administración que más se utiliza la sublingual, en estampillas que se aplican por algunos minutos de idéntica forma que LSD. La duración total de acción varía de 4 a 10 horas por vía sublingual.
Decenas de muertes y hospitalizaciones se han producido en Estados Unidos por el consumo de estos nuevos compuestos sintéticos, donde se describe que la sobredosis provoca taquicardia, hipertensión, hipokalemia, exacerbación de los síndromes serotoninérgico y simpaticomimético (4), temblores, agitación que pueden incluso llegar a durar en algunos casos hasta por tres días (5). Se han reportado casos de hipertermia y rabdomiólisis con un notable aumento de los valores plasmáticos de creatinkinasa y transaminasas (6), acidosis metabólica, falla renal, hepática y coagulación intravascular diseminada. En caso de intoxicaciones agudas y por consumo vía sublingual en estampillas se detectaría el compuesto original al no presentar efecto de metabolización por primer paso hepático (5).
Se presenta un método rápido de extracción en fase sólida (SPE, del inglés: solid-phase extraction) para análisis de 25C-NBOMe en orina como diagnóstico de intoxicación aguda, mediante cromatografía en capa delgada de alta resolución (HPTLC, del inglés: high-performance thin-layer chromatography) y la confirmación por cromatografía de gases/espectrometría de masa (CG/ EM), el cual puede convertirse en una herramienta de screening y confirmación rápida y confiable para detectar 25C-NBOMe en laboratorios forenses.
Materiales y Métodos
REACTIVOS
Metanol, acetato de etilo, acetonitrilo, amoníaco, dietilamina, tolueno, ácido acético, ácido clorhídrico 0,1N, diclorometano, isopropanol y ciclohexano, todos calidad cromatografía Merck (Darmstadt, Alemania).
Material de referencia de 25C-NBOMe gentilmente donado por la Drug Enforcement Administration (DEA, Washington).
Columnas de extracción en fase sólida SPE UCT CSDAU Clean Screen ® ácido octil-benzil-sulfónico 50 mg/3 mL CSDAU1L3 (Merck, Chile).
EQUIPOS
Cromatógrafo planar instrumental HPTLC equipado con Autosampler ATS4, equipos de desarrollo automático de placas ADC2, Espectro densitómetro Scanner 3, manejados por software Wincats, CAMAG (Muttenz, Suiza)
Cromatógrafo de gases Agilent 6890 N acoplado a detector de espectrometría de masas 5973B con autosampler de 100 posiciones manejado por software de adquisición de datos Chemstation Agilent (California, Estados Unidos)
Condiciones Instrumentales:
Condiciones cromatográficas de HPTLC
Aplicación de estándares y muestras: Las muestras se aplicaron en volumen de 25 μL en bandas de 5 mm, mediante equipo ATS-4, utilizando una jeringa de 100 μL y nitrógeno como gas para formar el spray de aplicación. La distancia entre cada banda (track) fue de 5,8 milímetros. Los estándares (2,0 μL) y muestras se aplicaron en placas de 20 x 10 cm, previamente acondicionadas a 80 °C por 20 minutos.
Desarrollo cromatográfico: Una vez aplicadas las muestras en la placa, ésta se desarrolla en la cámara de desarrollo automático ADC-2. Con un tiempo de pre acondicionado de 2,0 min, se utilizaron 10 mL de fase móvil por cada placa y la distancia de desarrollo fue de 70 milímetros. La fase móvil empleada consiste en una mezcla de Ciclohexano/ Tolueno/Dietilamina, 75/15/10 en volumen.
Detección instrumental: Una vez desarrollada la placa se escanea en el espectro densitómetro TLC scanner 3, la dimensión del slit fue de 4,00 x 0,30 mm, la velocidad de escaneo fue de 20 mm/segundo; la mínima área considerada para la integración fue de 5UA. Una vez obtenidas las bandas de cada analito se obtiene el espectro Ultravioleta de 190-400 nm, a partir del cual se seleccionó la longitud de onda de 298 nm. Los densitogramas se evaluaron con el software Wincats®, con el fin de confirmar el análisis, se tomó el espectro ultravioleta de cada banda del analito de interés de 190-400 nm y se comparó con los espectros de referencia creados a partir del estándar de 25C-NBOMe y almacenados en la biblioteca del equipo.
Confirmación de identidad de muestras incautadas
Para la confirmación de la identidad se aplicó la técnica de cromatografía de gases con detector selectivo de masas (CG/EM); las condiciones cromatográficas seleccionadas fueron las siguientes:
Inyector: Splitless 5 μL, temperatura 250 °C
Horno: Temperatura inicial 75 °C por 1 minuto. Luego aumentar 25 °C por minuto hasta 280 °C mantener por 20 minutos
Columna: HP-5 EM flujo Helio 1,0 mL por minuto
Detector: EM modo SIM, iones 121, 91 y 150
PROCESAMIENTO DE LAS MUESTRAS
La matriz seleccionada es la orina debido a la fácil obtención y volumen disponible, además, publicaciones acerca de análisis de derivados de NBOMe emplean orina como matriz de elección ya que debido a la vía de administración de la estampilla (sublingual) se detectará el compuesto como tal, sin metabolizar (7). Los pasos involucrados en la extracción de la matriz orina consisten en:
1. Volumen de muestra: 2 mL de orina + 2 mL de buffer fosfato 100 mM, pH 6, luego centrifugar por 5 minutos a 2000 rpm.
Acondicionar la columna de extracción en fase sólida (SPE) con 1 mL de metanol (tres veces), seguido de 1 mL de agua desionizada (tres veces) y tres veces con 1 mL de buffer fosfato pH 6 100 mM (aspirar al vacío).
2. Cargar la muestra en el cartucho de extracción SPE dispuesto en sistema Manifold (Presión 5 psi, flujo aproximado 1- 2 mL/minuto)
3. Lavar tres veces con 1 mL de agua desionizada, seguido de tres veces con 1 mL ácido acético 100 mM y posteriormente tres veces con metanol 1 mL; secar al vacío por 5 minutos.
4. Eluir tres veces con 1000 μL de mezcla de diclorometano/isopropanol/amoníaco, 78/20/2 en volumen (preparada en el día).
Agregar 50 μL de una solución al 1% de ácido clorhídrico en metanol y evaporar a sequedad en corriente de nitrógeno a 40 °C.
Dejar enfriar y reconstituir en 200 μL de metanol grado HPLC e inyectar en CG/EM (5 μL) y en cromatografía en capa delgada de alta resolución (HPTLC) (sembrar 25 μL).
VALIDACIÓN DEL MÉTODO
La validación de la metodología se realizó según los parámetros de la SWGTOX (8). Los análisis estadísticos para todos los parámetros de validación se efectuaron mediante el uso del software Statgraphics Centurion XVI ®.
ESPECIFICIDAD E INTERFERENCIAS
La selectividad de un método se investiga mediante el estudio de su capacidad para medir el analito de interés en muestras a las cuales deliberadamente se han introducido interferencias específicas de las que se cree que probablemente estén presentes en las muestras (9). Se estudió el efecto de la matriz orina mediante el uso de curvas de calibración en matriz orina blanco, libre de 25C-NBOMe.
LÍMITE DE DETECCIÓN Y CUANTIFICACIÓN LOD Y LOQ
El límite de detección (LOD) se define como la concentración mínima que puede distinguirse del ruido de fondo con un determinado grado de confianza. El límite de cuantificación (LOQ) es la concentración más baja del analito que puede ser determinada con un nivel aceptable de precisión. (10). Dentro de las múltiples formas de calcular el límite de detección y cuantificación se encuentran las siguientes fórmulas (11):
LOD = 3.3*Sa/a LOQ = 10* Sa/a
Donde Sa es el error total de la calibración y a es la pendiente de la curva de calibración (12).
PRECISIÓN
La precisión refleja los errores que se producen cuando se utiliza un método; las condiciones en que se mide la precisión se dividen en condiciones repetibles (Repetibilidad) y condiciones reproducibles (Reproducibilidad, también conocida como precisión intermedia). La precisión normalmente se mide en términos de coeficiente de variación (CV%) o desviación típica relativa (RSD). La precisión es expresada por el siguiente modelo:
![]()
Donde SD es la desviación estándar de las mediciones y –x corresponde a la media de los valores observados (8). Este parámetro se evaluó mediante la determinación de precisión intermedia en tres días distintos y en términos de repetibilidad analizando en un mismo día seis replicados de tres niveles distintos de concentración a 25, 50 y 100 ng/mL
LINEALIDAD
Se considera que un método es lineal cuando existe una relación directamente proporcional entre la respuesta obtenida y la concentración del analito en la matriz dentro del rango de concentraciones del analito buscado (rango de trabajo). Se elaboró una curva de calibración de seis niveles aplicados en triplicado en HPTLC, para evaluar la linealidad; frecuentemente se utiliza como criterio de la linealidad un coeficiente de correlación (r) elevado, del 0,99 o mejor (13).
EXACTITUD
Lo más utilizado es estimar la exactitud mediante el porcentaje de recuperación, analizando muestras añadidas con al menos tres concentraciones distintas (baja, media y alta) que abarquen la totalidad del rango de trabajo. La proporción entre los dos resultados (muestras añadidas y patrones de referencia) se denomina recuperación, generalmente referido como porcentaje de recuperación (%R). La exactitud se determina según la siguiente fórmula:
![]()
Donde la concentración encontrada promedio (C enc prom) corresponde a la media de los replicados y la concentración nominal (C Nominal) es la concentración agregada (fortificada o spike) del material de referencia. Para este parámetro se emplearon muestras fortificadas a los niveles de concentración esperados y reportados en la literatura sobre intoxicaciones, derivados NBOMes, que van de 28 a 36 ng/mL en orina para el caso de intoxicaciones agudas producto del consumo de estampillas vía oral de 25C-NBOMe (14)(15), según lo anterior y como además no se dispone de niveles de corte Cut-off se pretende llegar al límite de detección más bajo posible, con adecuada precisión y exactitud (13), para este propósito, se prepararon fortificados de orina a tres niveles 25, 50 y 100 ng/mL para evaluar la exactitud del método por HPTLC expresado como porcentaje de recuperación. Las muestras descritas se inyectaron además en CG/EM modo SIM para comprobar la identidad.
ROBUSTEZ
La robustez del método por HPTLC se estableció mediante la evaluación del impacto en la selectividad y capacidad de separación de distintas composiciones de fase móvil. Para esto se aplicó el método con tres distintas fases móviles y posteriormente se evaluaron los Rf y los espectros UV obtenidos para el analito en cada fase móvil, evaluando si estas composiciones pueden afectar la correcta identificación de muestras y estándares. Además, se emplearon distintos lotes y marcas de cromatoplacas.
Resultados
El método de extracción propuesto fue evaluado en términos de interferencias de la matriz, linealidad, límites (LOD y LOQ), precisión en condiciones de repetibilidad y reproducibilidad y el porcentaje de recuperación, obteniéndose los resultados que se muestran en la Tabla I.
Tabla I. Resultados de implementación de metodología por HPTLC para 25-C-NBOMe en Orina.

La Figura 1 muestra la identificación del analito 25C-NBOMe por HPTLC en orina y la comparación con los Rf y espectros UV de los estándares (track 1 al 10) destaca que en todos los casos la comparación es satisfactoria con una correlación mayor a 0,99. La Figura 2 denota el análisis de una muestra de orina fortificada a 25 ng/mL analizada por CG/EM y un estándar a 100 ng/mL.

Figura 1. Comparación de muestras fortificadas de orina y estándares de 25-C-NBOMe.
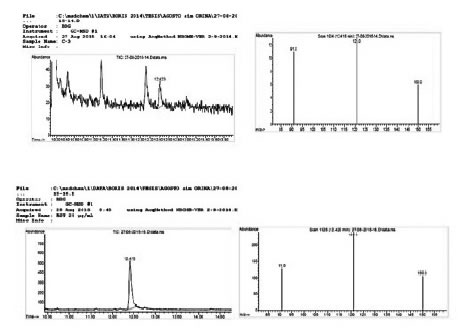
Figura 2. Cromatograma modo SIM
Discusión y Conclusiones
El método consiste en una extracción en fase sólida mediante columnas Clean Screen CSDAU® con relleno de ácido octil-bencil-sulfónico. Para la determinación instrumental se empleó el método por HPTLC y la confirmación por CG/EM en modo de monitoreo selectivo de iones SIM (iones m/z 121, 91, 150). El método propuesto permite detectar la presencia de 25C-NBOMe en orina a partir de 25 ng/mL con adecuada exactitud y precisión, con un mínimo uso de solventes y en corto tiempo. El sistema de extracción Manifold permite además procesar hasta 20 muestras de forma simultánea; al evaporar el extracto y reconstituirlo a 200 μL permite alcanzar adecuados niveles de detección instrumental, de ser necesario este extracto podría ser reconstituido a menor volumen mejorando la sensibilidad del método. La solución empleada para la elución de los analitos retenidos en el cartucho de extracción en fase sólida debe ser preparada en el momento ya que se observó que este aspecto va en directa relación con la eficiencia de la extracción. El método presenta adecuada exactitud, la precisión puede ser mejorada a niveles menores a los 50 ng/mL, ya que bajo este nivel se encuentra muy cercano al LOD del método, en tal caso se sugiere llevar el extracto final a menor volumen o sembrar mayor cantidad de muestra por banda. El método de detección por cromatografía de gases con detector selectivo de masas, permite detectar el analito sin interferencia de la matriz al tiempo de retención buscado y sin la necesidad de emplear derivatizante, lo que implica un menor costo en reactivos y en tiempo de análisis.
El método implementado consistió en un proceso de extracción en fase sólida con columnas que hasta la fecha no habían sido probadas para estos analitos. Estas columnas resultaron ser de gran utilidad y de capacidad de retención ya que se obtuvieron extractos limpios y sin interferencias de la matriz orina concentrando a un volumen de 200 μL sin la necesidad de derivatizar el extracto para su detección instrumental por HPTLC o CG/EM modo SIM. La validación resultó ser satisfactoria a los niveles de concentración estudiados (ng/mL o ppb) cumpliendo con los límites establecidos por la SWGTOX, obteniendo valores comparables a los valores informados en publicaciones internacionales. Lo anteriormente señalado permite que este método sea implementado en laboratorios forenses que requieran extraer NBOMes en orina para el diagnóstico de una intoxicación aguda y posterior detección instrumental.
1. Duffau B, Rojas S, Jofré S, Kogan M, Triviño I, Fuentes P. Thin-Layer Chromatography method for the detection of N,N-dimethyltryptamine in seized street samples. J Planar Chromatogr Mod TLC 2014; 27(6): 477–9.
2. Rickli A, Luethi D, Reinisch J, Buchy D, Hoener MC, Liechti ME. Receptor interaction profiles of novel N-2-methoxybenzyl (NBOMe) derivatives of 2,5-dimethoxy-substituted phenethylamines (2C drugs). Neuropharmacology 2015; 99: 546–53.
3. Duffau B, Camargo C, Cassels BK, Kogan M, Fuentes E. Analysis of a new potent hallucinogen, 25-B-NBOMe, in blotters by High-Performance Thin-Layer Chromatography. JPC - J Planar Chromatogr - Mod TLC. 2015; 28 (5): 395–7.
4. Rose SR, Poklis JL, Poklis A. A case of 25I-NBOMe (25-I) intoxication: a new potent 5-HT2A agonist designer drug. Clin Toxicol Philadelphia Pa 2013; 51 (3): 174–7.
5. Poklis JL, Devers KG, Arbefeville EF, Pearson JM, Houston E, Poklis A. Postmortem detection of 25I-NBOMe [2-(4-iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl)methyl]ethanamine] in fluids and tissues determined by high performance liquid chromatography with tandem mass spectrometry from a traumatic death. Forensic Sci Int 2014; 234: e14–20.
6. Hill S, Doris T, Gurung S. Severe clinical toxicity associated with analytically confirmed recreational use of 25I-NBOMe: case series. Clin Toxicol [Internet]. 2013; 51(May): 487–92.
7. Stellpflug SJ, Kealey SE, Hegarty CB, Janis GC. 2-(4-Iodo-2,5-dimethoxyphenyl)-N-[(2-methoxyphenyl) methyl]ethanamine (25I-NBOMe): Clinical Case with Unique Confirmatory Testing. J Med Toxicol 2014 Mar; 10(1): 45-50. [ Links ]
8. SWGTOX. Scientific Working Group for Forensic Toxicology (SWGTOX) Standard Practices for Method Validation in Forensic Toxicology. J Anal Toxicol 2013; 37: 452–74.
9. Peters FT, Drummer OH, Musshoff F. Validation of new methods. Forensic Sci Int 2007; 165 (2-3): 216–24.
10. Centro Nacional de Metrología. MÉTODOS ANALÍTICOS ADECUADOS A SU PROPÓSITO Guía de Laboratorio para la Validación de Métodos y Temas Relacionados. 2005. 1-69. [ Links ]
11. Renger B, Végh Z, Ferenczi-Fodor K. Validation of thin layer and high performance thin layer chromatographic methods. J Chromatogr A 2011; 1218 (19): 2712–21.
12. Srivastava M. High-performance thin-layer chromato-graphy (HPTLC). High-Performance Thin-Layer Chromatography (HPTLC). Springer Berlin Heidelberg 2011; 1-397. [ Links ]
13. Naciones Unidas. Directrices para la validación de métodos analíticos y la calibración del equipo utilizado para el análisis de drogas ilícitas en materiales incautados y especímenes biológicos. Viena: Sección de laboratorio y asuntos científicos Oficina de las Naciones Unidas contra la Droga y el Delito; 2010. 1–76.
14. Suzuki J, Dekker MA, Valenti ES, Arbelo Cruz F, Correa AM, Poklis JL, et al. Toxicities associated with NBOMe Ingestion, a Novel Class of Potent Hallucinogens: a Review of the Literature. Psychosomatics. 2015; 56 (2): 129–39.
15. Papoutsis I, Nikolaou P, Stefanidou M, Spiliopoulou C, Athanaselis S. 25B-NBOMe and its precursor 2C-B: modern trends and hidden dangers. Forensic Toxicol 2014; 1–11.
Recibido: 9 de noviembre de 2015.
Aceptado: 13 de enero de 2016.

