










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.50 no.2 La Plata jun. 2016
HEMOSTASIA Y TROMBOSIS
Factor von Willebrand y Enfermedad de von Willebrand: nuevos enfoques diagnósticos
Von Willebrand Factor and von Willebrand disease: new approaches to diagnosis
Fator Von Willebrand e doença de von Willebrand: novas abordagens diagnósticas
Adriana Inés Woods1a, Alicia Noemí Blanco1b, Ana Catalina Kempfer1a, Juvenal Paiva2b, Emilse Inés Bermejo1b, Analía Sánchez Luceros1,3a,b, María Angela Lazzari3a
1 PhD.
2 MSc.
3 MD.
a Laboratorio de Hemostasia y Trombosis, IMEX- CONICET- Academia Nacional de Medicina, Buenos Aires.
b Departamento de Hemostasia y Trombosis, Instituto de Investigaciones Hematológicas “Mariano R Castex”, Academia Nacional de Medicina, Buenos Aires, Argentina.
CORRESPONDENCIA DRA. ADRIANA I WOODS Pacheco de Melo 3081 (C1425AUM) CIUDAD AUTÓNOMA DE BUENOS AIRES, Argentina. E-mail: aiwoods@hematologia.anm.edu.ar
Resumen
El factor von Willebrand (VWF) es una glicoproteína que se sintetiza en células endoteliales y en megacariocitos. Su vida media es de ~12 horas. Está formado por multímeros de diferentes pesos moleculares, pequeños, intermedios, grandes y extragrandes. La actividad funcional reside en los multímeros grandes, y los extragrandes son trombogénicos. Promueve la adhesión plaquetaria al subendotelio, la agregación plaquetaria y transporta al FVIII en plasma, protegiéndolo de su degradación por proteasas. La enfermedad de von Willebrand es el trastorno hemorrágico más frecuente; se describen deficiencias cuantitativas (parcial: VWD1; total: VWD3) o defectos cualitativos (VWD2A, VWD2M, VWD2B y VWD2N). La expresión clínica es variable (sangrado muco-cutáneo) y su herencia autosómica, dominante o recesiva, según las variantes. Los niveles del VWF dependen de factores genéticos y no genéticos que afectan el diagnóstico y la expresión clínica. Para llegar al diagnóstico se precisan varias pruebas, algunas inespecíficas. El laboratorio comienza con pruebas orientadoras, se continúa con pruebas confirmatorias, y posteriormente pruebas para definir la variante de VWD. El diagnóstico genotípico es fundamental para lograr el diagnóstico diferencial entre VWD2B vs. PT-VWD y VWD2N vs. Hemofilia A (leve-moderada), diferenciar VWD de AVWS y discriminar variantes VWD2.
Palabras clave: Factor von Willebrand; Enfermedad de von Willebrand; Fenotipo; Genotipo.
Summary
Von Willebrand factor (VWF) is a glycoprotein with essential roles in both primary and secondary hemostasis, synthesized by endothelial cells and megakaryocytes. Its half-life is ~12 hours. VWF consists in multimers of different molecular weight: small, intermediate, large and ultra large. The functional activity resides in the large multimers; the ultra large are thrombogenic. VWF promotes platelet adhesion to subendothelium, platelet aggregation and binds FVIII, protecting it from proteolysis and preserving its hemostatic function. Von Willebrand disease is the most common bleeding disorder; qualitative defects (VWD2A, VWD2M, VWD2B and VWD2N) and quantitative deficiencies (VWD1 and VWD3) are described. The clinical expression is variable (mucocutaneous bleeding); VWF levels depend on genetic and non-genetic factors affecting diagnosis and clinical expression. The inheritance can be autosomal, dominant or recessive according to the variants. To reach diagnosis, several tests are required, being some of them unspecific. The laboratory testing begins with global tests, followed by confirmatory tests and further tests to define the variant of VWD. Genotypic studies are essential to achieve the differential diagnosis between VWD2B vs. PT-VWD, VWD2N vs. Hemophilia A (mild to moderate) and differentiate VWD from AVWS and discriminate VWD2 variants.
Keywords: Von Willebrand factor; Von Willebrand disease; Phenotypic diagnosis; Genotypic diagnosis.
Resumo
O fator de von Willebrand (vWF) é uma glicoproteína sintetizada em células endoteliais e em megacariócitos. Sua vida média é de ~12 horas. É constituído por multímeros de pesos moleculares diferentes, pequenos, intermediários, grandes e extragrandes. A atividade funcional reside nos multímeros grandes, sendo os extragrandes, trombogênicos. Promove adesão das plaquetas ao subendotélio, a agregação plaquetária e transporta o FVIII em plasma, protegendo-o de sua degradação. A doença de von Willebrand é o distúrbio hemorrágico mais frequente; são descritas deficiências quantitativas (parcial: VWD1; total: VWD3) ou defeitos qualitativos (VWD2A, VWD2M, VWD2B e VWD2N). A expressão clínica é variável, (sangramento mucocutâneo), e sua herança autossômica dominante ou recessiva de acordo com as variantes. Os níveis de vWF dependem de fatores genéticos e não-genéticos que afetam o diagnóstico e a expressão clínica. Para fazer o diagnóstico, vários testes são necessários, alguns inespecíficos. O laboratório começa com testes orientadores, continua com testes de confirmação e, mais tarde, com testes para definir a variante de VWD. O diagnóstico genotípico é essencial para alcançar o diagnóstico diferencial entre VWD2B vs. PT-VWD e VWD2N vs. Hemofilia A (leve a moderada), diferenciar VWD de AVWS, discriminar variantes VWD2.
Palavras chaves: Fator de von Willebrand; Doença de von Willebrand; Fenótipo; Genótipo.
Introducción
La enfermedad de von Willebrand (VWD) es el defecto hemorrágico congénito más frecuente. La prevalencia estimada varía según se considere el número de pacientes sintomáticos registrados en centros de hemofilia (0,0023-0,06%) o los estudios poblacionales (0,6-1,3%), dependiendo también de los criterios diagnósticos utilizados (1). Además de los defectos congénitos, hay formas adquiridas (AVWS), cuya prevalencia se estima en 0,04% (1). Mucho de lo que se sabe actualmente sobre la VWD surgió en la década de 1970, a partir de los primeros ensayos para determinar el antígeno del factor von Willebrand (VWF:Ag); pero fue la clonación del factor VIII (FVIII) y del factor von Willebrand (VWF) en la década de 1980, lo que permitió evidenciar la diferencia genética entre ambos factores y dar las bases moleculares para la comprensión del defecto. Se produce por la deficiencia y/o defecto del VWF, glicoproteína multimérica que circula en el plasma unida al FVIII de la coagulación, formando un complejo (FVIII-VWF) (2). Su modo de herencia es autosómico. La molécula del VWF contiene varios sitios funcionales de unión a ligandos; es capaz de unirse a receptores plaquetarios como la glicoproteínas Ib (GPIb) y IIbIIIa (GPIIb-IIIa), a componentes de la matriz subendotelial como el colágeno y al FVIII (3). Es útil tener presente las abreviaturas aceptadas internacionalmente para denominar a las proteínas y sus funciones (Tabla I). El defecto en el VWF puede causar sangrado al afectar la función plaquetaria o al reducir la concentración de FVIII. La relación entre los niveles de VWF y los síntomas clínicos o el riesgo de sangrado no han sido totalmente elucidados (4). Se describen 6 tipos diferentes de VWD debidos a deficiencia cuantitativa parcial (tipo 1) o total (tipo 3) del VWF o caracterizados por diferentes defectos cualitativos (tipo 2: 2A, 2B, 2M, 2N) (5).
Tabla I. VWF. Abreviaturas utilizadas

Al momento del diagnóstico es preciso tener presente que se trata de una enfermedad heterogénea, reflejo de la compleja estructura del VWF, así como de la influencia de factores adicionales (no debidos al gen VWF), que condicionan la presentación clínica de la enfermedad (3). El interrogatorio médico es fundamental para establecer la historia personal y familiar de sangrado (3). Además, se debe realizar una apropiada investigación de laboratorio para definir el fenotipo (3). El análisis genotípico se aplica a algunas situaciones puntuales (3). Se propone considerar la respuesta a la administración de desmopresina (DDAVP) como un elemento diagnóstico adicional (4). Se resumirán las características del VWF que faciliten la comprensión de la enfermedad y su clasificación. Se discutirá la interpretación y utilidad de los elementos diagnósticos disponibles, poniendo especial énfasis en los enfoques fenotípicos y genotípicos.
Rol del factor von Willebrand
El proceso fisiológico que limita la extravasación de la sangre luego de una lesión endotelial se conoce como hemostasia, y se caracteriza por dos fases claramente diferenciadas: Primaria: lleva a la formación del tapón plaquetario en el sitio de la lesión endotelial. Implica la presencia de plaquetas y del VWF en el sitio de la injuria. Secundaria: involucra proteínas plasmáticas (factores de la coagulación) en una serie de reacciones (cascada de la coagulación) que conducen a la formación del coágulo. El FVIII participa como cofactor del proceso; su sobrevida depende de su unión al VWF (evita la degradación por proteasas), el cual lo transporta al sitio de la lesión. Además, disminuye la inmunogenicidad de los concentrados de FVIII/VWF y protege al FVIII de su neutralización por anticuerpos.
La importancia de estas reacciones se evidencia en las manifestaciones clínicas hemorrágicas de los individuos con deficiencias (FVIII: hemofilia A, FIX: hemo- filia B, VWF: VWD). También desempeña un rol en la trombosis y un factor de riesgo en enfermedad coronaria (6), aún cuando la elevación (>200 UI/dL) sea por exceso de medicación para controlar el sangrado (7).
Origen, estructura y síntesis del VWF
El VWF es una glicoproteína de alto peso molecular que se sintetiza en células endoteliales (CE) y en la progenie megacarioblástica. El gen que lo codifica está localizado en el brazo corto del cromosoma 12, involucra 178 kb de ADN genómico organizado en 52 exones). Hay un pseudogen en el cromosoma 22, cuya función se desconoce, que posee 97% de homología con los exones 23 y 34 del gen VWF (8).
La estructura primaria del VWF está constituida por un monómero, que a través de sucesivos pasos de dimerización y multimerización, forma una serie de multímeros de diferente peso molecular: pequeños (LMWM), intermedios (IMWM), grandes (HMWM) y extra grandes (ULMWM). Los HMWM son los que poseen la mayor actividad funcional. Los ULMWM son más activos, pero circulan en plasma en niveles muy reducidos y transitoriamente (9). El VWF se sintetiza inicialmente en el retículo endoplásmico (RE) como un precursor (pre-pro-VWF) (Fig. 1). Al separarse el péptido señal del extremo amino-terminal, el pro-VWF se ensambla formando dímeros a través de uniones de puentes disulfuro (S-S) entre los extremos carboxi-terminal (dominio CK). Finalmente, después de sucesivos procesos de sulfatación, glicosilación, multimerización entre los extremos amino-terminal de los dímeros y clivaje proteolítico del VWFpp (Fig. 2), el VWF y el VWFpp se secretan al plasma en cantidades equimolares. Se almacena como ULMWM o se libera directamente al plasma, siendo posteriormente degradado a multímeros más pequeños y menos trombóticos (10).

Figura 1. Estructura del VWF
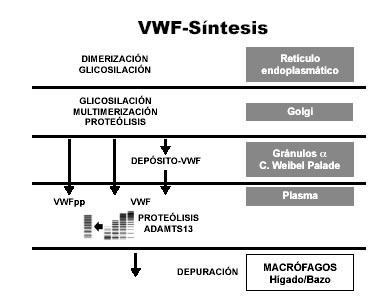
Figura 2. Síntesis del VWF.
Hay dos sitios de depósito:
– los cuerpos de Weibel-Palade (WPB) de la CE,
– los gránulos á de los megacariocitos y dos vías de secreción:
– constitutiva,
– regulada.
Predomina la constitutiva, con liberación continua del VWF al plasma y a la matriz subendotelial. El 85% del VWF plasmático proviene de las CE; sólo 5-10% es secretado por la vía regulada (11), en respuesta a estímulos fisiológicos o patológicos. La secreción desde los gránulos á (vía regulada) es secundaria a la activación plaquetaria por agonistas. La vida media del VWF en plasma es de 8-12 horas (2) y la del VWFpp, 2-3 horas (4).
Dominios del VWF
El pre-pro-VWF está compuesto por diferentes dominios repetitivos y un dominio cystein-knot (CK) en el extremo C-terminal (Fig. 1). Por microscopía electrónica, los dominios D se observan como lóbulos formados por ensamble de módulos (D y D4N, 8 cisteínas “C8”, trypsin inhibitor-like “TIL” y fibronectin type 1 like “E”) (12).
Los sitios funcionales, que involucran la unión a múltiples ligandos (colágeno, heparina, ristocetina, glicoproteínas plaquetarias GPIb y GPIIb-IIIa), se encuentran localizados en los diferentes dominios (Fig. 3): -D’/parte D3 unión a FVIII, -A1 unión a GPIbá y a colágeno VI y IV, -A2 sitio catalítico de ADAMTS13, -A3 unión a colágeno I y III, -C4 (secuencia RGDS: Arg-Gly- Asp-Ser) unión a GPIIb-IIIa (plaquetas activadas) (3). A través del dominio A3, el VWF se une al colágeno subendotelial expuesto por el daño vascular; el alto shear stress generado por el flujo, cambia la conformación del VWF inmovilizado y expone el dominio A1, reclutando plaquetas por unión a la GPIbá, además del sitio proteolítico del dominio A2. El shear stress es esencial en la activación y regulación del VWF; constituye un refinado mecanismo para impedir la formación de coágulos en sitios no deseados (13).

Figura 3. Sitios de unión a ligandos en los dominios del VWF.
La función del VWF depende de la formación de los HMWM; los residuos Cys, involucrados en la formación de puentes S-S inter- o intra-cadenas (dimerización o multimerización) son importantes para su función, por lo que mutaciones que involucren a estos residuos tendrán consecuencias que se verán reflejadas en la calidad y/o cantidad de la proteína resultante (14).
Proteólisis de VWF
ADAMTS13. Los ULVWF son los más activos
Plasmina. La acción de la plasmina sobre el VWF es un mecanismo dependiente del shear stress y de las plaquetas (15); degrada rápidamente los complejos plaquetas- VWF sobre la superficie de las CE activadas, en ausencia de ADAMTS13.
Proteasas leucocitarias. Las proteasas de neutrófilos y calpaínas atacan los HMWM generando fragmentos diferentes a los presentes habitualmente en plasma (176 kDa y 140 kDa). Las proteasas podrían ser fisiológicamente importantes para regular el tamaño de los multímeros (15).
Modificadores de los niveles de VWF Los valores de referencia plasmáticos de VWF son muy amplios (0,4 to 2,4 UI/mL). Los niveles bajos están relacionados con sangrado. Los síntomas de sangrado, además de inespecíficos, son frecuentes en la población general, por lo que es clave diferenciar entre individuos sanos y portadores de VWD. El nivel en plasma es producto de la relación entre producción y depuración.
Entre los factores modificadores, el grupo sanguíneo es uno de los más conocidos y estudiados. También influyen la edad, así como factores raciales y hormonales o polimorfismos en el gen VWF.
Grupo sanguíneo ABO. Individuos grupo O tienen niveles más bajos de FVIII:C y/o VWF. El VWF está sujeto a N- y O-glicosilación y transporta antígenos del grupo ABO (A, B y H), siendo el perfil de glicosilación uno de los determinantes de su sobrevida en circulación. Es motivo de debate el uso valores de referencia ABO-específicos; dado que el fenotipo de sangrado se asocia a los niveles de VWF, independiente del grupo sanguíneo, se sugiere no usar rangos ABO-específicos (16).
Edad. Es otro factor que implica diferencias significativas en los niveles de VWF; a partir de los 40 años habría un aumento permanente del VWF plasmático (17). Factores raciales. En mujeres afroamericanas se han descripto niveles más elevados (18).
Factores hormonales. Hay discrepancias respecto de las variaciones durante el ciclo menstrual. Se sugiere repetir los estudios de mujeres sin diagnóstico, preferentemente en esa fase. En el embarazo, el VWF y el FVIII aumentan durante todo el período gestacional y el puerperio inmediato, alcanzando 2-3 veces los valores basales, volviendo a los valores pre-embarazo en pocas semanas (19). El incremento es suficiente para corregir deficiencias cuantitativas parciales pero no cualitativas ni severas (20).
Depuración de VWF
Hay varias vías de eliminación (depuración o clearance) del VWF de la circulación: se reconocen dos receptores que parecen intervenir en la remoción del VWF de la circulación: el CLEC4M sobre las células endoteliales y Siglec-5 en los macrófagos y LRP-1 en macrófagos y hepatocitos (21). Diversas mutaciones se asocian al aumento de la depuración, en VWD1, VWD1C (incluye Vicenza), en VWD2A y VWD2B. Se ha observado aumento de la depuración post- DDAVP en p.Arg1315Cys, p.Arg1379Cys y p.Lys1794Glu (22). La relación VWFpp/VWF:Ag en plasma ayuda a identificar el mecanismo fisiopatológico, prediciendo probablemente el tipo de respuesta al DDAVP (23).
Polimorfismos del gen VWF
El VWF es altamente polimórfico. La Sociedad Internacional de Hemostasia y Trombosis (ISTH), en su base de datos (www.vwf.group.shef.ac.uk), recopila 181 polimorfismos (SNP) diferentes, algunos afectan las determinaciones de laboratorio, sin afectar el fenotipo (24). Otro estudio mostró 2728 SNPs y 91 inserciones/deleciones en el gen VWF, con mayor variabilidad étnica en africanos y asiáticos (25). Otros SNP se asociaron a aumento de la proteolisis por ADAMTS13 o con resistencia a la proteolisis (26).
Enfermedad de von Willebrand Fue descrita por Erick von Willebrand, en una familia procedente de las islas Åland-Finlandia. El modo de herencia es autosómica e irregular: dominante (tipo 1, 2A, 2B y 2M) y recesiva (tipo 3 y 2N). Hay descriptas mutaciones de novo, resultado de la aparición de una mutación nueva en una célula germinal de los padres (óvulo o espermatozoide) o en el zigoto. Hay también defectos adquiridos (síndrome de von Willebrand adquirido: AVWS), frecuentemente no reconocidos o erróneamente clasificados como VWD. (27).
Síntomas clínicos
La expresión clínica puede ser severa, moderada o leve, según el defecto en el VWF o los niveles del FVIII:C y puede diagnosticarse en la primera infancia, la adolescencia o la edad adulta. Presenta sangrado muco-cutáneo (epistaxis, gingivorragias, hematomas fáciles, equimosis, sangrado post exodoncia, sangrado prolongado ante trauma mínimo, sangrado excesivo postoperatorio) y en las mujeres meno-metrorragia, sangrado post parto y post cesárea, folículos hemorrágicos en el momento de la ovulación; a diferencia de la hemofilia que se caracteriza por hemartrosis y hemorragias musculares o cerebrales. Se han diseñado cuestionarios para dar una puntuación según los síntomas, denominados bleeding score (BS), como herramienta para cuantificar las manifestaciones y contribuir a discriminar entre sujetos con y sin VWD1 (28), con VWD tipos 2 y 3 (29), VWD2B (30) y un cuestionario (PBQ) y score pediátricos (31). El “bleeding rate” es el número de episodios hemorrágicos que ocurren durante un período de tiempo definido; podría usarse como valor pronóstico y ayudar a identificar como sangradores a pacientes sin historia de hemorragias mayores (32). Para definir menorragia, se utiliza el Pictorial Bleeding Assessment Chart (PBAC) (33).
Descripción de anormalidades del VWF en la VWD
Las anormalidades pueden ser:
• Interacción anormal entre las plaquetas: tipo VWD2B (aumento de la interacción con la GPIbá), tipos VWD2A y VWD2M (disminución de la interacción con la GPIbá).
• Defecto en la interacción con el colágeno: tipos VWD2M, VWD2A y VWD2B.
• Depuración anormal en el VWD1C (disminución de la vida media) y en los VWD2A y VWD2B.
• Defecto en la interacción con el FVIII en el tipo VWD2N.
• Defectos en la dimerización y/o multimerización, alteración del transporte intracelular y secreción o proteólisis aumentada.
Variantes de VWD-Fenotipo
Es causada por defectos cuantitativos y cualitativos del VWF. Según los mecanismos fisiopatológicos (Tabla II) se agrupan en:
Tabla II. Clasificación de la VWD.

Defectos Cuantitativos
• Disminución de la síntesis del VWF: tipos VWD1 (deficiencia parcial) y VWD3 (deficiencia total).
• Disminución marcada del VWF, pueden detectarse multímeros extragrandes del VWF con sobrevida del VWF acortada (depuración anormal): VWD1C (incluye al Vicenza).
Tipo 1. Se caracteriza por VWF:RCo/VWF:Ag>0,6. Comprende el 70-80% de los pacientes con VWD. El patrón multimérico es normal; la concentración en plasma es variable (10-40 UI/dL), con valores normales o disminuidos de VWF en plaquetas. Hay disminución de la síntesis del VWF y descenso secundario del FVIII. Según los niveles plasmáticos se habla de VWD1 severo, definido y probable VWD1 (34) (Tabla III). Las mutaciones responsables se localizan a lo largo de todo el gen, aunque la posibilidad de hallar mutaciones sólo es probable en pacientes con VWF<30 UI/dL.
Tabla III. Diferencias en las pruebas de laboratorio entre variantes cuantitativas.

Tipo 1C. Representa el 10-15% de los VWD1. Se caracteriza por aumento de la depuración del VWF (vida media: <3 h). La disminución de VWF (15-30 UI/dL) se acompaña de aumento del VWFpp/VWF:Ag. Responde a la infusión de DDAVP, pero los niveles de VWF declinan rápidamente. El patrón multimérico muestra formas extra grandes o bandas satélites disminuidas.
Tipo 3. Es el menos frecuente (1/106, según los grupos étnicos) y con síntomas clínicos más severos, VWF:RCo y VWF:Ag no detectables y FVIII:C muy bajo (<10 UI/ dL). Genotípicamente se caracteriza mayoritariamente por la presencia de grandes deleciones del gen, aunque también se han descripto mutaciones ins/del, lo que produce un cambio del marco de lectura, que lleva a la aparición de un codón de terminación, con pérdida o disminución de síntesis de la proteína. Tanto las deleciones como las mutaciones se localizan a lo largo de todo el gen.
Defectos Cualitativos (defectos en la interacción del VWF con diferentes ligandos)
Tipo 2. Corresponde al 15-20% de los pacientes. Con concentraciones de VWF:Ag y de FVIII:C normales (>50 UI/dL), o disminuidas, hay discordancia entre actividad y cantidad de proteína, subdivididas en 2A, 2B, 2M y 2N (5). Los 2A, 2B y 2M se caracterizan por VWF:RCo/ VWF:Ag<0,6 y el 2N por FVIII:C/VWF:Ag<0,7 (Tabla IV).
Tabla IV. Diferencias en las pruebas de laboratorio entre variantes cualitativas.
VWD2A. Caracterizada por mutaciones en su mayoría localizadas en el dominio A2 (¡Ö73%), que alteran la dimerización o multimerización del VWF, con disminución o ausencia de los HMWM e IMWM en plasma y plaquetas. Se traduce en disminución o ausencia de la agregación plaquetaria inducida con ristocetina (RIPA), disminución o ausencia de VWF:RCo y VWF:CB, normal o ligera disminución de VWF:Ag y FVIII:C (35).
VWD2M. Caracterizada por mutaciones en su mayoría en el dominio A1 (¡Ö76%) que generan incapacidad de unión a la GPIbá. La estructura multimérica es normal.
Hay pérdida de la función del VWF, con VWF:RCo disminuido o ausente, con VWF:Ag y FVIII:C normales o levemente disminuidos. El VWF:CB suele ser normal, aunque depende del tipo de colágeno utilizado en el ensayo. El subtipo 2CB (mutaciones en el dominio A3) con deficiencia VWF:CB, pero con VWF:Ag, VWF:RCo y composición multimérica normales, se incluye en el VWD2M (36). Estos casos estarían sub diagnosticados, dado que no siempre se incluye el VWF:CB en el panel de evaluación.
VWD2B. Todas las mutaciones responsables se localizan en el dominio A1. Estas producen aumento de la afinidad del VWF por la GPIbá (5) (ganancia de función), detectada por el ensayo de RIPA a bajas dosis (¡Ü0,7 mg/mL), donde el VWF anómalo es capaz de aglutinar plaquetas a concentraciones subumbrales de ristocetina (37). Hay ausencia de HMWM en plasma; el VWF liberado se une a las plaquetas y es eliminado de la circulación, lo que puede producir trombocitopenia, (30). Factores fisiológicos (edad sexo, grupo sanguíneo), ejercicio o situaciones de estrés (embarazo, infecciones, cirugías) pueden agravar la trombocitopenia y el cuadro clínico. Los mecanismos fisiopatológicos son dos:
– agregación plaquetaria, seguida de remoción, llevando a trombocitopenia y pérdida de HMWM y
– bloqueo de la GPIbá y pérdida de la capacidad de adhesión al subendotelio.
La p.V1316M altera señales plaquetarias y conduce a inhibición de la GPIIb-IIIa; la trombocitopatía resultante podría contribuir a la tendencia hemorrágica. Se ha descripto VWD2B atípico con perfil multimérico y recuento de plaquetas normal (38). VWD2N. Hay alteración en la capacidad del VWF de unir FVIII, por mutaciones en los dominios D’-D3. Se refleja en disminución muy marcada del FVIII:C, por disminución de su vida media, con VWF:Ag, VWF:RCo y patrón multimérico normales. La prevalencia de los subtipos en nuestra casuística es: 69,6%, posible VWD1; 13,5%, VWD1 definido; 0,35%, VWD1 severo; 0,55%, VWD3; 9,5%, VWD2A; 0,6%, VWD2B; 2,5%, VWD2M y 3,4%, VWD2N (34). En la Figura 4 se muestra la localización más frecuente de las mutaciones responsables de las diferentes variantes.

Figura 4. Localización más frecuente de las mutaciones responsables de las distintas variantes de la VWD.
Anticuerpos anti-VWF
Se ha descripto la presencia de inhibidores en pacientes poli transfundidos con VWD3 (39) con una frecuencia de 5-10% (40). En algunos casos, la baja recuperación del VWF y/o la rápida depuración luego de la infusión de concentrados es el único indicador de la presencia de un anticuerpo. Pueden ser neutralizantes (inhiben al VWF) o no neutralizantes (forman complejos sin inhibir la función, pueden aumentar la depuración). También se han descripto autoanticuerpos. La mayoría de estos anticuerpos precipitan el VWF en plasma normal.
Síndrome de von Willebrand adquirido (AVWS)
Frecuentemente no es reconocido o se lo considera erróneamente como VWD. Se caracteriza por defecto del VWF secundario a síndromes linfoproliferativos (48%) o mieloproliferativos (15%), enfermedades cardiovasculares (21%; 40-46%) o malignas (5%), desórdenes autoinmunes (2%) o menos frecuentemente, hipotiroidismo, uremia; en algunos casos no se detecta ningún desorden de base. Los posibles mecanismos fisiopatológicos son:
– depuración aumentada mediada por anticuerpos, -inhibición de función mediada por anticuerpos,
– degradación por proteólisis aumentada, -adsorción (tumores, enfermedad de Gaucher, etc.) (41).
El diagnóstico es complejo y difícil (dada la heterogeneidad de mecanismos subyacentes), lo cual complica el manejo terapéutico (41). Algunos AVWS tienen anticuerpos que aumentan la depuración del VWF infundido (41). En VWD3 también pueden desarrollarse anticuerpos que inhiben al VWF infundido (42).
Diagnóstico de laboratorio de VWD
La evaluación clínica es fundamental para establecer la historia personal y familiar de sangrado. Esto se combina con una apropiada investigación de laboratorio para definir el fenotipo (39); en algunos casos la respuesta a la administración de desmopresina (DDAVP), utilizada en la terapéutica, puede ser un elemento diagnóstico adicional (43). El análisis genético contribuye en algunas situaciones definidas a definir el diagnóstico (39). Se discutirá la interpretación y utilidad de los elementos diagnósticos disponibles desde ambos enfoques: fenotípico y genotípico.
Enfoque Fenotípico
Estrategia Diagnóstica. Cuando la clínica resulta compatible con VWD, se impone efectuar estudios de laboratorio a fin de detectar el defecto y definir el fenotipo. La Figura 5 resume la estrategia sugerida (43), que consiste en:
– determinar la historia personal y familiar de sangrado.
– realizar pruebas “generales” de “screening”, que no confirman ni excluyen VWD
– evaluar el VWF (funcional e inmunológico) y el FVIII para diagnosticar VWD
– realizar pruebas complementarias para tipificar y clasificar el VWD (43), de importancia en el manejo clínico-terapéutico del paciente.
Las formas severas son más fáciles de diagnosticar. Dada la variabilidad ya mencionada, se impone repetir y confirmar los hallazgos en momentos diferentes, considerando las numerosas variables (ejercicio físico, cigarrillo, estrés, diabetes, hipotiroidismo, etc.) que afectan los resultados de las pruebas de laboratorio, en especial en individuos con clínica leve. También se evaluará la necesidad o importancia de estudiar a otros miembros de la familia.

Figura 5. Estrategia diagnóstica para estudiar pacientes con sintomatología hemorrágica
Pruebas utilizadas en la evaluación del fenotipo Las pruebas se dividen en screening o pesquisa, diagnósticas (FVIII/VWF), confirmatorias (clasificación) y otras. El algoritmo diagnóstico (Fig. 6) integra los resultados de las pruebas, para permitir la clasificación de los pacientes en los diferentes tipos de VWD.

Figura 6. Algoritmo diagnóstico.
Pruebas “generales” de screening (pesquisa)
Comprenden la evaluación de la función (tiempo de sangría, PFA-100) y número de plaquetas (recuento de plaquetas), así como de las diferentes vías del mecanismo de coagulación (TTPA; TP; TT). Hay discusión respecto a la utilidad del tiempo de sangría. Como objeciones se plantea que es invasivo, no-específico y poco reproducible (operador, presión del manguito, localización/dirección/ profundidad del corte, llanto y/o movimiento) (44). Se correlaciona bien con los síntomas clínicos; el resultado es variable según el tipo de VWD (muy prolongado: tipo severo y defectos cualitativos 2A/2B/2M); prolongado o normal: tipo 1. Los métodos más aceptados son: Ivy y Mielke (más sensible). No hay consenso para incluir la determinación con PFA-100, aunque hay laboratorios que lo hacen. Es más sensible que el TS; aún no se conoce su especificidad; se necesitan más datos para su recomendación a pesar de que su uso se inició en 1985. Los resultados de tiempo de sangría y PFA-100 no siempre coinciden. El recuento de plaquetas es particularmente útil en la pesquisa del subtipo VWD 2B, donde puede estar disminuido. El TTPA, a pesar de utilizar reactivos sensibles, sólo se altera si hay disminución de FVIII (<40-50 UI/dL), secundaria a disminución del VWF (VWD3, VWD2N, algunos VWD1); por lo tanto es frecuentemente normal (44). Los estudios de mezclas con plasma normal permiten evidenciar inhibidores (34). La utilidad de estas pruebas es discutida, su valor reside en evidenciar otros desórdenes que pudieran ser responsables o contribuir al cuadro clínico del paciente. En individuos con clara sospecha clínica de VWD y/o historia familiar, se recomienda investigar directamente FVIII y VWF (43).
Pruebas diagnósticas-FVIII/VWF
La evaluación se realiza mediante: -estimación (inmunológica) de la concentración del VWF en plasma (VWF:Ag); -evaluación de la actividad (VWF:RCo, unión a GPIb, anticuerpos anti-Dominio A1); -determinación de la actividad de FVIII (FVIII:C), reflejo indirecto de la función del VWF.
– VWF:Ag
Se determina mediante técnicas inmunológicas. Los métodos de elección son enzimo-inmunoensayos (ELISA) (alta sensibilidad: 0-5 UI/dL) (45) o inmuno-turbidimétricos (LIA) (menor sensibilidad: 5-10 UI/dL). Ambos métodos son altamente reproducibles y tiene una imprecisión del 5-15%. El método por citometría de flujo es bueno pero no está al alcance de muchos laboratorios (46).
– ACTIVIDAD DE VWF VWF:RCo.
Refleja la interacción del VWF con la GPIb/IX/V. Es la prueba elegida para evaluar la actividad del VWF. Se basa en la propiedad del VWF de aglutinar plaquetas en presencia de ristocetina in vitro y cuantificando la aglutinación producida. Alteraciones en los HMWM o mutaciones en el dominio A1 del VWF conducen a alteraciones en los niveles de VWF:RCo. Es difícil de estandarizar, tiene alto coeficiente de variación intra e inter laboratorio (10-40%), sobre todo en valores <15 UI/dL y tiene baja sensibilidad en niveles <10 UI/dL. Hay polimorfismos (p.Asp1472His), que pueden disminuir la unión a la ristocetina y otros (p.Ala1381Thr) tienen mayor afinidad por la GPIbá, por lo que dan valores artificialmente bajos/altos de VWF:RCo (47). Los métodos automatizados disminuyen la variación intra e inter ensayo, pero no la sensibilidad, dando resultados más elevados (48). En pacientes con bajo VWF:RCo (<10UI/dL) y sospecha clínica de inhibidor, se debe repetir el ensayo en mezclas con plasma normal para descartar su presencia mediante (34). VWF:Actividad (VWF:Act). Se basa en técnicas de ELISA o LIA (o quimioluminiscencia de mayor sensibilidad que LIA), utiliza anticuerpos monoclonales anti-VWF o anti-GPIb que reconocen el sitio de unión a la GPIb (dominio A1) o al VWF respectivamente. Son técnicas más reproducibles y sencillas que el VWF:RCo, pero es discutida su capacidad y utilidad para detectar alteraciones estructurales o funcionales el VWF. Estos métodos deben ser refrendados a través de estudios en pacientes con VWD tipo 2.
– FVIII:C
Mide la actividad coagulante. En el contexto de la evaluación de VWD, es reflejo indirecto de la capacidad del VWF de unir FVIII y estabilizar su vida media. La metodología más utilizada es el método en “una etapa” (automatizado, semi-automatizado o manual) (44). También es posible determinarlo mediante sustratos cromogénicos (44). Puede existir discordancia entre ambos métodos (defectos moleculares, inhibidores).
– COCIENTES
Los resultados de actividad de VWF deben ser analizados junto con los niveles de VWF:Ag y FVIII:C para una mejor interpretación diagnóstica. En individuos normales, la relación FVIII/VWF (FVIII/VWF:Ag; FVIII/ VWF:RCo o FVIII/VWF:CB) es ¡Ö1. Lo mismo ocurre para VWF:RCo/VWF:Ag y VWF:CB/VWF:Ag, donde valores <0,6 sugieren la presencia de VWF disfuncional (44); sin embargo, la variabilidad del VWF:RCo afecta su utilidad. Un cociente FVIII/VWF:Ag <0,6 sugiere VWD2N o hemofilia (49). Sería importante para los laboratorios contar con valores de corte propios, considerando la metodología aplicada.
Pruebas complementarias - Clasificación
Incluyen Multímeros, RIPA, VWF:CB, VWF:VIIIB y VWFpp
– MULTÍMEROS
El análisis de la concentración relativa de multímeros de diferente tamaño es una prueba, compleja y laboriosa. Consiste en la electroforesis de plasma o lisado plaquetario en geles de SDS-agarosa, con posterior identi- ficación de los multímeros con anticuerpos anti VWF, usando el complejo avidina-biotina peroxidasa u otras técnicas (47). Puede ser cualitativa o cuantitativa por densitometría. Según las características del gel, puede ser de “baja resolución” (1%) donde se observa todo el perfil multimérico (diferencia multímeros grandes de intermedios y pequeños) o “alta resolución” (1,7%) donde no se observan los multímeros grandes y se distingue la estructura de los tripletes (44). En general, el estudio se inicia a baja resolución, para detectar ausencia de multímeros grandes. La alta resolución evidencia defectos por aumento o disminución de proteólisis, así como defectos por aumento de depuración (5). Es crucial para distinguir 2M de 2A y 2B y útil para diagnosticar algunas variantes, en especial del tipo 2.
– AGREGACIÓN PLAQUETARIA INDUCIDA POR RISTOCETINA (RIPA)
Se realiza sobre plasma rico en plaquetas utilizando diferentes concentraciones de ristocetina; la agregación depende del VWF plasmático y de la GPIbá de las plaquetas (47). En ausencia “virtual” de VWF (VWD3) la agregación está muy disminuida o ausente (ristocetina <1,2 mg/mL); pudiéndose aumentar la dosis (1,5-2 mg/ mL) para “forzar” la respuesta (34). Mutaciones de la GPIb pueden dar RIPA ausente (Síndrome de Bernard-Soulier), o aumentada (von Willebrand plaquetario PT-VWD).
– RIPA-BAJAS DOSIS (RIPAbd)
Se induce agregación con concentraciones sub-umbrales de ristocetina (<0,7 mg/mL) que no producen agregación en individuos normales, pero sí en pacientes con ganancia de función del VWF (VWD2B) o mutaciones en la GPIbá (PT-VWD). La prueba es crítica para detectar VWD2B (43). Sólo 69% de los VWD2B tienen RIPA positiva a 0,5 mg/mL (50), concentración internacionalmente recomendada para la pesquisa de VWD2B (51); es importante buscar la concentración umbral y no descartar como posible VWD2B pacientes con fenotipo de sangrado con RIPAbd positivo sin trombocitopenia ni VWF:RCo/VWF:Ag anormal, especialmente en el caso de mujeres.
– UNIÓN VWF-COLÁGENO (VWF:CB)
Mide la capacidad del VWF plasmático de unirse al colágeno, principalmente a través del sitio de unión en el domino A3 (47). El método de detección es ELISA (47) o como alternativa, con alta sensibilidad, citometría de flujo. La reacción es dependiente de la estructura multimérica, las formas grandes tienen mayor avidez por el colágeno; además, influye el origen y tipo del colágeno. Puede distinguir VWD2M con mutaciones en los dominios A1 o A3; el colágeno tipo I-III tiene alta afinidad por el dominio A3 del VWF y menor afinidad por el dominio A1; los colágenos tipo IV y VI se unen específicamente al dominio A1 (52). En pacientes con síntomas hemorrágicos sin alteración de VWF:Ag y VWF:RCo, se deben considerar posibles mutaciones en A3, detectadas utilizando colágeno I-III. No hay consenso respecto a incluir o no el VWF:CB (43) junto al VWF:RCo en el estudio inicial de VWD; su importancia radicaría en detectar defectos en la unión al colágeno, con VWF:RCo normal (44).
– UNIÓN VWF-FVIII (VWF:FVIIIB)
Evalúa la habilidad del VWF a unirse a FVIII normal (exógeno) (47). Se determina por ELISA; se ensaya cuando el FVIII:C/Ag<0,7, para diferenciar VWD2N de hemofilia A. Presenta problemas de estandardización (falta estándares y de control de calidad externo) y reproducibilidad (falsos positivos) (43).
– PROPÉPTIDO VWF (VWFpp)
Se determina por ELISA usando anticuerpos de captura contra el VWFpp. Su nivel plasmático es proporcional al VWF:Ag y refleja la síntesis del VWF. El aumento de la relación VWFpp/VWF:Ag (>2) indica mayor depuración del VWF causada por diferentes mecanismos (4) (tipo 1C-Vicenza, AVWS) y predice una sobrevida acortada post-administración de DDAVP (4). En las Tablas III y IV se resumen los resultados de las pruebas de laboratorio en cada una de las variantes de la enfermedad.
Otras pruebas
– ANTICUERPOS ANTI-VWF
Los ensayos para detectar anticuerpos anti-VWF no están claramente establecidos. Es posible evidenciarlos mediante pruebas de corrección con plasma normal en los ensayos de VWF:RCo, VWF:CB, VWF:Ag y/o FVIII. El efecto no es tiempo-temperatura dependiente, el título se establece mediante una modificación del método Bethesda (53). Resultados negativos de los ensayos de mezcla no excluyen la presencia de un anticuerpo, dado que el mismo puede estar dirigido contra epitopes no funcionales; éstos pueden ser detectados por métodos de ELISA (sensibles, pero con presencia de falsos positivos). Se recomienda realizar la búsqueda de anticuerpos por ambos métodos: -ensayos de corrección con normal y -ELISA (40).
– DIAGNÓSTICO DIFERENCIAL ENTRE VWD 2B Y PT-VWD
Es obligatorio, dado que el tratamiento médico es diferente; sin embargo, no es sencillo, habiéndose reportado PT-VWD en 15% de casos inicialmente considerados VWD2B. Se deben realizar análisis de mezclas (54) y la agregación con crio precipitado o concentrado de VWF. Ambas pruebas son orientadoras. El diagnóstico definitivo se realiza mediante análisis genotípico (búsqueda de mutaciones en el exón 28 del gen VWF y en el gen GP1BA). Respuesta al DDAVP del paciente: Como prueba adicional se pueden tener en cuenta los datos que provienen de la misma. Como se muestra en las Tablas III y IV, el perfil de laboratorio observado dependerá del tipo de VWD.
Consideraciones pre-analíticas
Es fundamental un adecuado proceso pre-analítico (55). Defectos en la toma (inadecuada punción o relación anticoagulante/plasma, exceso de aplicación del torniquete), separación, transporte y/o almacenamiento de la muestra pueden dar resultados falsos. El FVIII es sensible tanto a la activación por trazas de trombina como a la degradación, favorecida por cambios de pH, etc. Puede haber alteraciones del VWF con, por ejemplo, pérdida de la estructura multimérica. El complejo FVIII-VWF es sensible a múltiples estímulos o situaciones fisiopatológicas (ejercicio, estrés, drogas, cambios hormonales, embarazo) que pueden modificar los niveles circulantes y llevar a interpretaciones erróneas de los resultados si no son tenidos en cuenta. Evitar situaciones de estrés, en especial en niños. No se recomienda estudiar a menores de 6 meses de edad. Durante el ciclo menstrual se sugiere realizar los estudios preferentemente durante los tres primeros días. El embarazo no es momento adecuado para estudiar pacientes con VWD (excepto ante la sospecha de variantes severas) y mucho menos, realizar la prueba de desmopresina.
Valores de referencia
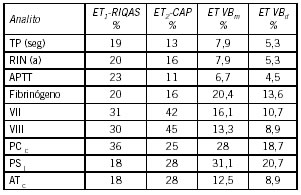
Es importante determinar para cada prueba, el valor de referencia en la población estudiada, con la metodología aplicada en cada laboratorio. FVIII, VWF:Ag, VWF:RCo, VWF:CB, VWFpp se calibran frente al estándar internacional 07/316 del National Institute of Biological Standards and Control (NIBSC), Londres. Como se mencionó anteriormente, no serían necesarios rangos para cada grupo ABO; el nivel de VWF condicionaría el riesgo de sangrado (44). No se utilizan valores de referencia por grupo etario, pero es importante recordar que VWF y FVIII aumentan con la edad.
Enfoque Genotípico
El análisis genético es fundamental para confirmar el diagnóstico de VWD2B vs. PT-VWD y VWD2N vs. Hemofilia A (leve-moderada), o diferenciar VWD de AVWS, permite identificar variantes de VWD2 (2A, 2B, 2M y 2N) localizadas, en general, en regiones determinadas del ADN complementario (ADNc) (34); también puede contribuir al consejo genético y prenatal o predecir el desarrollo de inhibidores en VWD3 (43). Desafortunadamente, el gran tamaño del gen VWF y la alta frecuencia de variaciones (SNP) no patológicas limita la utilidad del diagnóstico genético. No todos los pacientes tienen mutaciones identificables, en especial los VWD1, donde la presencia de mutaciones sería más común en pacientes con VWF:Ag<20-39 UI/dL. La situación es compleja, al no haberse establecido claramente la relación genotipo-fenotipo (39). La probabilidad de detectar el defecto genético es baja y requiere una evaluación exhaustiva y costosa, que contrasta con la baja utilidad clínica en la mayoría de los casos. Sin embargo, lo novedoso en este campo es su contribución a la resolución de casos de difícil diagnóstico, lo cual probablemente arroje una nueva luz para simplificar la actual clasificación de la VWD.
Estudio genotípico
El análisis genotípico se realiza mediante la secuenciación directa (Método de Sanger) de los fragmentos de interés, previamente amplificados por técnicas de PCR. Luego de la secuenciación, se compara el electroferograma del paciente frente a la secuencia normal (55). Se pueden utilizar métodos de screening como el Conformation Sensitive Gel Electrophoresis (CSGE) que permite el chequeo rápido de varios exones y pacientes. Si bien es sencillo y económico, hay que validar el método mediante la secuenciación del exón de interés.
Análisis de predicción in sílico
Las herramientas de predicción in sílico están siendo usadas cada vez más. El objetivo de estos programas es identificar el daño producido o no por un cambio de nucleótido y cómo este cambio afecta o no la estructura y función de la proteína en estudio, y por ende el fenotipo. Son una herramienta muy útil para la caracterización de nuevas sustituciones nucleotídicas, cuando los estudios de expresión son difíciles de realizar. Los más difundidos son: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) y SIFT (http://sift.bii.a-star.edu.sg/).
Análisis de alineamiento
Frente a la presencia de cambios nucleotídicos no previamente descriptos, se realiza el alineamiento del exón afectado del VWF humano con el de otras especies. Si la posición en estudio se corresponde con un residuo altamente conservado a lo largo del árbol filogenético, es altamente improbable que dicha posición tolere sustituciones sin cambios en las características funcionales del VWF. Existen diversas aplicaciones informáticas, como UniProt KB (http://www.uniprot.org).
Casos
Para dar una visión concreta de la importancia y aplicación del enfoque genotípico en el diagnóstico de VWD se discutirán algunos casos.
1- Diagnóstico diferencial VWD2B vs. PT-VWD
Paciente (varón, 23 años) con fenotipo de sangrado importante (BS: 11), macro trombocitopenia, RIPA bd positivo a 0,3 mg/mL de ristocetina y VWF:RCo/ VWF:Ag<0,2, compatible con VWD2B. Sin embargo, el estudio diferencial de mezclas y desafío con crio precipitados era compatible con PT-VWD. Se estudió el exón 28 tanto en el paciente como en su madre, no encontrando mutaciones. En el exón 2 del gen GP1BA se detectó una sustitución G>T localizada en la posición 738 del c.ADN, resultando en un cambio de aminoácido en el residuo 246 (p.Trp246Leu). La madre no es portadora de la mutación; el padre no pudo estudiarse (no reside en el país). El análisis in silico determinó que el cambio afecta la integridad de la proteína; el cambio no se localizó en los 100 alelos normales estudiados. Se confirmó el diagnóstico de PTVWF mediante el estudio genético del paciente (57). El ejemplo muestra la importancia del estudio genético en la diferenciación de ambos tipos de VWD.
2- Inusual hallazgo en las pruebas de laboratorio en una paciente embarazada con VWD2B
Paciente (27 años) (BS: 6) que se estudia en la semana 10 de embarazo: TS>9 min; FVIII:C=56 UI/ dL; VWF:Ag=47 UI/dL; VWF:RCo<10 UI/dL; RCo/ Ag=0,21; VWF:CB=6 UI/dL; CB/Ag<0,13; 150.000 plaquetas/109L; RIPA 1,2 mg/mL normal; RIPAbd 0,5 mg/mL ausente; ausencia de HMWM. Semana 26: TS=7 min; FVIII:C=90 UI/dL; VWF:Ag=64 UI/ dL; VWF:RCo=23 UI/dL; RCo/Ag=0,36; VWFpp/ Ag=2,08; 114.000 plaquetas/109L; RIPA 0,5 y 0,4 mg/ mL positivos. Dado que es infrecuente la presencia de VWF:RCo<10 UI/dL en pacientes con VWD2B (30), los resultados iniciales sugerían un fenotipo VWD2A. Sin embargo, fue sorprendente hallar VWF:RCo<10 UI/ dL con RIPA 1,2 mg/mL normal, y recuento de plaquetas en el límite; lo que sugería un fenotipo VWD2B. El RIPA 0,5 mg/mL ausente en el 1º estudio podría relacionarse al VWF:RCo<10 UI/dL. En un 2º estudio, el aumento del VWF:RCo por el embarazo indujo trombocitopenia y positivizó el RIPAbd a 0,5 y 0,4 mg/mL. Se pensó que en esta paciente, se requerió un mínimo nivel de VWF:RCo para positivizar RIPAbd. Se procedió a realizar el análisis genético, detectándose la mutación p.Arg1308Cys) en heterocigosis, confirmando el diagnóstico de VWD2B. La positivización del RIPA y la trombocitopenia observados con la progresión del embarazo y la genotipificación fueron decisivos para confirmar el diagnóstico (34).
3- Mutaciones de novo
Se halló la p.Y1542D en una niña (2 años) con fenotipo de sangrado (BS: 2) y diagnóstico fenotípico de VWD2A. Su hermana y padres eran asintomáticos; los estudios de laboratorio fueron compatibles con fenotipo normal y no presentaban en el análisis genético la mutación hallada en el propósito. La Tabla V muestra el haplotipo del propósito y de su padre. Confirmada la paternidad, se determina la presencia de la mutación de novo.
Tabla V. Haplotipo de la niña con la p.Y1542D y su padre.

Conclusiones
Dada la complejidad de las bases moleculares, no es extraño que el diagnóstico de VWD también lo sea.
Enfoque fenotípico
La expresión del VWF y su concentración en plasma es variable, dependiendo de factores genéticos y no genéticos; ello puede enmascarar el defecto y complicar el diagnóstico. Hay dificultades metodológicas y pre-analíticas que deben considerarse. Los problemas pueden ser minimizados mejorando los métodos de detección, usando métodos sensibles y precisos, estimando valores de referencia propios, ampliando el panel de pruebas para evaluar el fenotipo, evitando estudiar al paciente en condiciones de estrés, post-ejercicio, enfermedad, infección, embarazo, tratamiento hormonal, corticoides, etc. Hay que confirmar los resultados, considerar datos adicionales como la respuesta al DDAVP en el análisis final.
Enfoque genotípico
Si bien el diagnóstico y la clasificación no requieren en general del análisis genético hay situaciones especia- Factor y enfermedad de von Willebrand les donde la genotipificación es mandatoria para lograr el diagnóstico diferencial entre VWD2N vs. Hemofilia A o VWD2B vs. PT-VWD, entre VWD2M y VWD1 severo; puede también contribuir al consejo genético y mejorar el manejo clínico (VWD3). La nuevas metodologías, como los estudios in sílico, facilitan su aplicación en la resolución de casos complejos, donde el fenotipo pueda inducir a un error diagnóstico o no permita caracterizar adecuadamente el defecto. La correcta clasificación tiene implicancias terapéuticas, como administrar concentrados de VWF en lugar de concentrados de FVIII (VWD2N vs. hemofilia) o concentrados de VWF en lugar de reemplazo de plaquetas (VWD2B vs. PT-VWD). Hay un camino a recorrer antes de contar con pruebas accesibles que permitan la caracterización inequívoca (fenotípica y genotípica) del VWD. Es necesario aclarar que hasta ahora, no hay ninguna prueba de laboratorio, fenotípica o genotípica que se relacione como marcador de riesgo de sangrado.
1. Laffan MA, Lester W, O’Donnell JS, Will A, Tait RC, Goodeve A, et al. The diagnosis and management of von Willebrand disease: a United Kingdom Haemophilia Centre Doctors Organization guideline approved by the British Committee for Standards in Haematology. Br J Haematol 2014 Nov; 167 (4): 453-5.
2. Terraube V, O’Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 2010; 16: 3-13.
3. Lillicrap D. von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013 Nov 28; 122 (23): 3735-40. [ Links ]
4. Castaman G, Rodeghiero F. Advances in the diagnosis and management of type 1 von Willebrand disease. Expert Rev Hematol 2011; 4: 95-106. [ Links ]
5. Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor J Thromb Haemost 2006; 4: 2103-4. [ Links ]
6. Sonneveld MA, Cheng JM, Oemrawsingh RM, de Maat MP, Kardys I, Garcia-Garcia HM, et al. Von Willebrand factor in relation to coronary plaque characteristics and cardiovascular outcome. Results of the ATHEROREMO- IVUS study. Thromb Haemost 2015; 113: 577-84. [ Links ]
7. Lazzari MA, Sanchez-Luceros A, Woods AI, Alberto MF, Meschengieser SS. Von Willebrand factor (VWF) as a risk factor for bleeding and thrombosis. Hematology 2012; 17 Suppl 1: S150-2. [ Links ]
8. Mancuso DJ, Tuley EA, Westfield LA, Lester-Mancuso TL, Le Beau MM, Sorace JM, et al. Human von Willebrand factor gene and pseudogene: structural analysis and differentiation by polymerase chain reaction. Biochemistry 1991; 30: 253-69. [ Links ]
9. Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, et al. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood 2002; 99: 3971-7. [ Links ]
10. Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002; 100: 4033-9. [ Links ]
11. Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large biologically potent von Willebrand factor multimers. Cell 1986; 46: 185-90. [ Links ]
12. Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood 2012; 120: 449-58. [ Links ]
13. Interlandi G, Ling M, Tu AY, Chung DW, Thomas WE. Structural basis of type 2A von Willebrand disease investigated by molecular dynamics simulations and experiments. PLoS One 2012; 7: e45207. [ Links ]
14. Woods AI, Sanchez-Luceros A, Kempfer AC, Powazniak Y, Calderazzo Pereyra JC, Blanco AN, et al. C1272F: a novel type 2A von Willebrand’s disease mutation in A1 domain; its clinical significance. Haemophilia 2012; 18: 112-6.
15. Wohner N, Kovács A, Machovich R, Kolev K. Modulation of the von Willebrand factor-dependent platelet adhesion through alternative proteolytic pathways. Thromb Res Suppl Apr 2012; 129: e41-6. [ Links ]
16. De Wee EM, Sanders YV, Mauser-Bunschoten EP, van der Bom JG, Degenaar-Dujardin ME, Eikenboom J, et al; for the WiN study group. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thromb Haemost 2012; 108: 683-92. [ Links ]
17. Davies JA, Hathaway LS, Collins PW, Bowen DJ. von Willebrand factor: demographics of plasma protein level in a large blood donor cohort from South Wales in the United Kingdom. Haemophilia 2012; 18: e79-81. [ Links ]
18. Miller CH, Haff E, Platt SJ, Rawlins P, Drews CD, Dilley AB, et al. Measurement of von Willebrand factor activity: relative effects of ABO blood type and race. J Thromb Haemost 2003; 1: 2191-7. [ Links ]
19. Sánchez-Luceros A, Meschengieser SS, Marchese C, Votta R, Casais P, Woods AI, et al. Factor VIII and von Willebrand factor changes during normal pregnancy and puerperium. Blood Coagul Fibrinolysis 2003; 14: 647-51. [ Links ]
20. Lee CA, Chi C, Pavord SR, Bolton-Maggs PH, Pollard D, Hinchcliffe-Wood A, et al. UK Haemophilia Centre Doctors’ Organization. The obstetric and gynaecological management of women with inherited bleeding disorders-- review with guidelines produced by a taskforce of UK Haemophilia Centre Doctors’ Organization. Haemophilia 2006; 12: 301-36.
21. Casari C, Lenting PJ, Wohner N, Christophe OD, Denis CV. Clearance of von Willebrand factor. J Thromb Haemost 2013; 11 Suppl 1: 202-11. [ Links ]
22. Castaman G, Lethagen S, Federici AB, Tosetto A, Goodeve A, Budde U, et al. Response to desmopressin is influenced by the genotype and phenotype in type 1 von Willebrand disease (VWD): results from the European Study MCMDM-1VWD. Blood 2008; 111: 3531-9. [ Links ]
23. Castaman G. New development in von Willebrand disease. Curr Opin Hematol 2013; 20: 424-9. [ Links ]
24. Flood VH, Gill JC, Morateck PA, Christopherson PA, Friedman KD, Haberichter SL, et al. Common VWF exon 28 polymorphisms in African Americans affecting the VWF activity assay by ristocetin cofactor. Blood 2010; 116: 280-6. [ Links ]
25. Wang QY, Song J, Gibbs RA, Boerwinkle E, Dong JF, Yu FL. Characterizing polymorphisms and allelic diversity of von Willebrand factor gene in the 1000 Genomes. J Thromb Haemost 2013; 11: 261-9. [ Links ]
26. Pruss CM, Notley CR, Hegadorn CA, O’Brien LA, Lillicrap D. ADAMTS13 cleavage efficiency is altered by mutagenic and, to a lesser extent, polymorphic sequence changes in the A1 and A2 domains of von Willebrand factor. Br J Haematol 2008; 143: 552-8.
27. Tiede A. Diagnosis and treatment of acquired von Willebrand syndrome. Thromb Res 2012; 130 Suppl 2: S2-6. [ Links ]
28. Rodeghiero F, Castaman G, Tosetto A, Batlle J, Baudo F, Cappelletti A, et al. The discriminant power of bleeding history for the diagnosis of type 1 von Willebrand disease: an international, multicenter study. J Thromb Haemost 2005; 3:2619-26. Erratum in: J Thromb Haemost 2006; 4: 925. [ Links ]
29. Gill JC, Christopherson PA, Flood VH, Friedman KD, Montgomery RR The Zimmerman Program Investigators. Bleeding Scores in Von Willebrand Disease (VWD) Re-Visited: Analysis of the TS Zimmerman Program for the Molecular and Clinical Biology of VWD. Blood (ASH Annual Meeting Abstracts), Nov 2008; 112: 425. [ Links ]
30. Federici AB, Mannucci PM, Castaman G, Baronciani L, Bucciarelli P, Canciani MT, et al. Clinical and molecular predictors of thrombocytopenia and risk of bleeding in patients with von Willebrand disease type 2B: a cohort study of 67 patients. Blood 2009; 113: 526-34. [ Links ]
31. Bowman M, Riddel J, Rand ML, Tosetto A, Silva M, James PD. Evaluation of the diagnostic utility for von Willebrand disease of a pediatric bleeding questionnaire. J Thromb Haemost 2009; 7: 1418-21. [ Links ]
32. Tosetto A, Castaman G, Rodeghiero F. Bleeders, bleeding rates, and bleeding score. J Thromb Haemost 2013; 11 Suppl 1: 142-50. [ Links ]
33. Janssen CA, Scholten PC, Heintz AP. A simple visual assessment technique to discriminate between menorrhagia and normal menstrual blood loss. Obstet Gynecol 1995; 85: 977-82. [ Links ]
34. Woods AI, Sánchez-Luceros A, Meschengieser SS, Kempfer AC, Blanco AN, Lazzari MA. Diagnosis and management of von Willebrand disease in a single institution of Argentina. Semin Thromb Hemost 2011; 37: 568-75. [ Links ]
35. O’Brien LA, Sutherland JJ, Weaver DF, Lillicrap D. Theoretical structural explanation for Group I and Group II, type 2A von Willebrand disease mutations. J Thromb Haemost 2005; 3: 796-7.
36. Keeling D, Beavis J, Marr R, Sukhu K, Bignell P. A family with type 2M VWD with normal VWF:RCo but reduced VWF:CB and a M1761K mutation in the A3 domain. Haemophilia 2012; 18: e33. [ Links ]
37. Federici AB, Canciani MT. Clinical and laboratory versus molecular markers for a correct classification of von Willebrand disease. Haematologica 2009; 94: 610-5. [ Links ]
38. Casonato A, Gallinaro L, Cattini MG, Pontara E, Padrini R, Bertomoro A, et al. Reduced survival of type 2B von Willebrand factor, irrespective of large multimer representation or thrombocytopenia. Haematologica 2010; 95: 1366-72. [ Links ]
39. Goodeve AC. The genetic basis of von Willebrand disease Blood Reviews 2010: 24: 123-34. [ Links ]
40. James PD, Lillicrap D, Mannucci PM. Alloantibodies in von Willebrand disease. Blood 2013; 122: 636-40. [ Links ]
41. Tiede A, Rand JH, Budde U, Ganser A, Federici AB. How I treat the acquired von Willebrand syndrome. Blood 2011; 117: 6777-85. [ Links ]
42. Collins P, Budde U, Rand JH, Federici AB, Kessler CM. Epidemiology and general guidelines of the management of acquired haemophilia and von Willebrand syndrome. Haemophilia 2008; 14 (Suppl.3): 49-55. [ Links ]
43. Favaloro EJ. Rethinking the diagnosis of von Willebrand disease. Thrombosis Research 2011:127 (Suppl. 2):S17-S21. [ Links ]
44. Nichols WL, Hultin MB, James AH, Manco-Johnson MJ, Montgomery RR, Ortel TL, et al. von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14: 171-232. [ Links ]
45. Taylor LD. The application of the biotin/avidin system to the von Willebrand factor antigen immunoassay. Thromb Haemost 1988; 59: 251-4. [ Links ]
46. Kempfer AC, Silaf MR, Farias CE, Carballo GA, Woods AI, Lazzari MA. Binding of von Willebrand factor to collagen by flow cytometry. Am J Clin Pathol 1999; 111: 418-23. [ Links ]
47. En: Fundamentos para el manejo práctico en el laboratorio de Hemostasia. Editores: Kordich L, Blanco A, Cerrato G, Quintana I, Vázquez A, Vizcargüénaga M, Grupo CAHT, segunda edición. La Plata: Federación Bioquímica de la Provincia de Buenos Aires, 2013. [ Links ]
48. Favaloro EJ, Bonar R, Marsden K. Lower limit of assay sensitivity: an under-recognised and significant problem in von Willebrand disease identification and classification. Clin Lab Sci 2008 Summer; 21: 178-83. [ Links ]
49. Castaman G, Montgomery RR, Meschengieser SS, Haberichter SL, Woods AI, Lazzari MA. von Willebrand’s disease diagnosis and laboratory issues. Haemophilia 2010; 16 (Suppl.5): 67-73.
50. Woods AI, Bermejo E, Sánchez-Luceros A, Kempfer AC, Calderazzo JC, Meschengieser SS, et al. Analysis of clinical phenotype and genotype in patients with VWD2B, in a single institution of Argentina. J Thromb Haemost 2013; 11 (Supl. 2): 944 (a. [ Links ] 3036).
51. Ozeki M, Kunishima S, Kasahara K, Funato M, Teramoto T, Kaneko H, et al. A family having type 2B von Willebrand disease with an R1306W mutation: Severe thrombocytopenia leads to the normalization of high molecular weight multimers. Thromb Res 2010; 125: e17-22. [ Links ]
52. Flood VH, Schlauderaff AC, Haberichter SL, Slobodianuk TL, Jacobi PM, Bellissimo DB, et al; Zimmerman Program Investigators. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood 2015; 125: 2297-304. [ Links ]
53. Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-Bunschoten E. The Nijmegen modification of the Bethesda assay for factor VIII:C inhibitors: improved specificity and reliability. Thromb Haemost 1995; 73: 247-51. [ Links ]
54. Favaloro EJ, Patterson D, Denholm A, Mead S, Gilbert A, Collins A, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from Type 2B VWD using a simplified ristocetin-induced-platelet- agglutination mixing assay and confirmed by genetic analysis. Br J Haematol 2007; 139: 623-6. [ Links ]
55. Lippi G, Plebani M, Favaloro EJ. Technological advances in the hemostasis laboratory. Semin Thromb Hemost 2014; 40: 178-85. [ Links ]
56. Mancuso DJ, Tuley EA, Westfield LA, Worrall NK, Shelton- Inloes BB, Sorace JM, et al. Structure of the gene for human von Willebrand factor. J Biol Chem 1989 25; 264: 19514-27. [ Links ]
57. Woods AI, Sanchez-Luceros A, Bermejo E, Paiva J, Alberto MF, Grosso SH, et al. Identification of p.W246L as a novel mutation in the GP1BA gene responsible for platelet-type von Willebrand disease. Semin Thromb Hemost 2014; 40: 151-60. [ Links ]
Recibido: 4 de mayo de 2016.
Aceptado: 10 de mayo de 2016.

