










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.50 no.4 La Plata dic. 2016
BIOQUÍMICA CLÍNICA
Metabolismo del hemo: las dos caras de los efectos de la acumulación de precursores y porfirinas
Heme metabolism: the janus nature of precursorsand porphyrins
Metabolismo do hemo: as duas faces dos efeitos da acumulação de precursores e porfirinas
María Victoria Rossetti1a*, Ana María Buzaleh1ab*, Victoria Estela Parera1a*, Haydée Fukuda1a*, María Elisa Lombardo1ab*, Jimena Lavandera1a***, Esther Noemí Gerez1a**, Viviana Alicia Melito1ab, Johanna Romina Zuccoli2a, Silvina Fernanda Ruspini2a, Vanesa Rocío Puente3a, Berenice Andrea Diez1a, María Julieta Teijo1a, Gabriela Nora Cerbino3a, Laura Sabina Varela3a, Marcelo Néstor Guolo1a y Alcira María del Carmen Batlle1,4a****
1 Doctor, UBA.
2 Licenciada en Biotecnología, UNQUI.
3 Licenciada en Biología, UBA.
4 Ph.D. in Sciences, London University.
a Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Hospital de Clínicas José de San Martín, Universidad de Buenos Aires (UBA); Argentina.
b Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires (UBA). Argentina.
* Investigador Principal de la Carrera del Investigador Científico CIC-CONICET.
** Investigadora Adjunta de la CIC-CONICET.
*** Investigadora Asistente de la CIC-CONICET.
**** Investigadora Superior de la CIC-CONICET.
CORRESPONDENCIA Prof. Dra. ALCIRA BATLLE Viamonte 188l 10º "A" C1056ABA - BUENOS AIRES - Argentina Tel: 54 11 4812 3357. Fax: 54 11 4811 7447 E-mail: batllealcira@yahoo.com.ar
Resumen
Las porfirias son enfermedades metabólicas consecuencia de fallas en la biosíntesis del hemo, caracterizadas por un patrón específico de acumulación y excreción de intermediarios, responsables de su patofisiología. En las porfirias agudas el exceso de ácido d-aminolevúlico (ALA) produce una sintomatología neuroabdominal asociada al daño oxidativo por formación de especies reactivas de oxígeno (ROS), originadas por autooxidaxión del ALA. En las cutáneas, la sintomatología es producto de la acumulación de porfirinas, que como el ALA, inducen la formación de ROS. Su desencadenamiento se precipita por factores endógenos (ayuno, estrés, hormonas) y/o exógenos (fármacos), en particular algunos anestésicos. Se presenta una revisión de los estudios bioquímicos y genéticos en pacientes con diferentes porfirias obtenidos en el Centro de Investigaciones de Porfirias y Porfirinas (CIPYP), durante los últimos 38 años, que permitieron ampliar el conocimiento sobre las bases moleculares sobre estas patologías. Se describen los logros resultantes del empleo de modelos experimentales de porfiria, inducida farmacológica o genéticamente, que contribuyeron a la clasificación de algunas drogas como prohibidas para pacientes con porfiria. Finalmente, las porfirinas generadoras de ROS, y por ende inductoras de muerte celular, tienen su aplicación para combatir infecciones por organismos hemo-deficientes como Trypanosoma cruzi y también para ser utilizadas como fotosensibilizadores en la terapia fotodinámica (TFD).
Palabras clave: Metabolismo del hemo; Porfirias; Porfirinas; Ácido d-aminolevúlico; Anestésicos volátiles; Terapia fotodinámica; Trypanosoma cruzi.
Summary
Porphyrias comprise a group of metabolic disorders of the heme biosynthesis pathway resulting in a specific accumulation and excretion of intermediates which are responsible for their pathophysiology. Acute porphyrias are characterized by acute neurovisceral symptoms due to the overproduction and accumulation of d-aminolevulinic acid (ALA) which leads to an oxidative damage resulting from the formation of reactive oxygen species (ROS). In cutaneous porphyrias, the symptomatology is a result of porphyrin accumulation which also induces ROS moulding. In both cases, their clinical signs are precipitated by endogenous factors (stress, hormones, low calories intake) and/or exogenous drugs, in particular some anaesthetics. A review of the biochemical and genetic results obtained from patients with different porphyrias, diagnosed at the CIPYP during the last 38 years is presented here, aimed at obtaining additional evidence about the molecular nature of these disorders. The achievements obtained from experimental porphyria models –pharmacologically or genetically induced– are also described, which contributed to the classification of some drugs as prohibited for their use in porphyric patients. Finally, as porphyrins produce ROS and therefore cellular death, they can be used to treat infections by heme-deficient organisms like Trypanosoma cruzi and also as photosensitizers in photodynamic therapy (TFD).
Key words: Heme metabolism; Porphyrias; Porphyrins; D-aminolevulinic acid; Volatile anaesthetics; Photodynamic therapy; Trypanosoma cruzi.
Resumo
As Porfirias são doenças metabólicas decorrentes de falhas na biossíntese do Hemo, caracterizadas por um padrão específico de acumulação e excreção de intermediários responsáveis de sua patofisiologia. Nas Porfirias Agudas, o excesso de ácido δ-aminolevulínico (ALA) produz uma sintomatologia neuroabdominal associada ao dano oxidativo por formação de espécies reativas de oxigênio (ROS), decorrentes da auto-oxidação do ALA. Nas Cutâneas a sintomatologia é produto da acumulação de porfirinas, que como o ALA, induzem a formação de ROS. Seu desencadeamento precipita-se por fatores endógenos (jejum, estresse, hormônios) e/ou exógenos (fármacos), especialmente alguns anestésicos. Apresenta-se uma revisão dos estudos bioquímicos e genéticos em pacientes com diferentes Porfirias obtidos no Centro de Investigações de Porfirias e Porfirinas (CIPYP), durante os últimos 38 anos, que permitiram ampliar o conhecimento sobre as bases moleculares destas patologias. Descrevem-se as conquistas resultantes do uso de modelos experimentais de Porfiria, induzida farmacológica ou geneticamente, que contribuíram à classificação de algumas drogas como proibidas para pacientes com Porfiria. Afinal, as porfirinas geradoras de ROS e, por conseguinte, indutoras de morte celular têm sua aplicação para combater infecções por organismos hemo-deficientes como Trypanosoma cruzi e também ser utilizadas como fotossensibilizadores na terapia fotodinâmica (TFD).
Palavras-chave: Metabolismo do hemo; Porfirias; Porfirinas; Ácido δ-aminolevulínico; Anestésicos voláteis; Terapia fotodinâmica; Trypanosoma cruzi.
Introducción
El hemo es el grupo prostético de varias hemoproteínas indispensables para el metabolismo celular. Se sintetiza a partir de moléculas simples como la glicina y succinilCoA, que dan origen al precursor ácido d-aminolevúlico (ALA), reacción catalizada por la enzima ALA sintetasa (ALA-S). A partir de dos moléculas de ALA y por acción del ALA dehidrasa (ALA-D) o PBG-sintetasa se forma el monopirrol porfobilinógeno (PBG). Cuatro moléculas de PBG se condensan por acción de la PBG deaminasa (PBG-D) o hidroximetilbilano (HMB) sintetasa para formar el tetrapirrol de cadena abierta, HMB, el cual se reordena y cicla al isómero fisiológico, uroporfirinógeno III (UROgen III), reacción catalizada por la uroporfirinógeno sintetasa o isomerasa. A partir de aquí, una serie de reacciones de decarboxilación y decarboxilación oxidativa, catalizadas por enzimas específicas, URO decarboxilasa (URO-D), coproporfirinogenasa (CPGasa) y protoporfirinógeno oxidasa (PPox), llevan a la formación de protoporfirina IX (PPIX), que por incorporación de una molécula de hierro catalizada por la enzima ferroquelatasa (FECH) sintetiza finalmente hemo (1) (Figura 1).
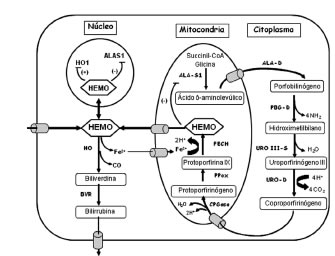
Figura 1. Metabolismo del hemo y su regulación en hígado.
ALA-S: Ácido d-aminolevúlico sintetasa, ALA-D: Ácido d-aminolevúlico dehidrasa, PBG-D: Porfobilinógeno deaminasa, UROgen III-S: Uroporfirinógeno III sintetasa, URO-D: Uroporfirinógeno decarboxilasa, CPGasa: Coproporfirinogenasa, PPox: Protoporfirinógeno oxidasa, FECH: Ferroquelatasa, HO: Hemo oxigenasa, BRV: Biliverdina reductasa.
La principal vía de degradación del hemo es catalizada por la enzima hemo oxigenasa (HO), obteniéndose CO, biliverdina e Fe2+; la biliverdina posteriormente es convertida a bilirrubina por la enzima biliverdina reductasa y transportada al hígado para formar bilirrubina conjugada y ser excretada del organismo (2)(3) (Figura 1). En humanos se han identificado 3 isoformas de HO: HO-1, HO-2 y HO-3; la HO-1 es inducible frente al estrés oxidativo, shock térmico, hipoxia, lipopolisacáridos, metales pesados, citoquinas y su propio sustrato (4).
La biosíntesis del hemo está finamente regulada de tal forma que en condiciones normales se acumulan y excretan mínimas de intermediarios. En el hígado, esta regulación se ejerce esencialmente por inhibición y represión del ALA-S (ALA-S 1) por el hemo. En eritrocitos, la mayor fuente de hemo para la síntesis de hemoglobina, la regulación está ejercida principalmente por la cantidad de Fe en la célula sobre la isoforma específica del ALA-S (ALA-S 2) y el receptor de transferrina, mediante un complejo mecanismo sobre la traducción de dichas proteínas (Figura 2).

Figura 2. Regulación de la síntesis de hemo en células eritroideas.
ALA-S: Ácido d-aminolevúlico sintetasa, Tf: Transferrina, Tf-r: Receptor de transferrina.
La deficiencia de algunas de las enzimas del camino del hemo, excepto la ALA-S, conduce a su desregulación con acumulación de los precursores ALA y PBG y/o porfirinas y a la expresión de enfermedades conocidas como porfirias. Sin embargo, cuando esta acumulación se induce por diferentes métodos puede tener aplicaciones sumamente ventajosas.
Este trabajo trata tanto de los aspectos negativos de la acumulación de precursores y/o porfirinas como de los efectos benéficos de su utilización con fines terapéuticos.
Porfirias
En la temática de las porfirias, el Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP) es un Instituto del CONICET y la UBA; desde 1978 es un centro de referencia único en Latinoamérica, dedicado desde hace 38 años al diagnóstico, tratamiento y seguimiento de los pacientes con distintos tipos de porfirias. El CIPYP posee reconocimiento internacional (www.orphan-europe.com), está avalado por numerosas publicaciones, incluyendo libros, que han contribuido al mejor conocimiento científico así como al desarrollo de nuevas terapias para el tratamiento de todas estas enfermedades, varias de ellas publicadas en Acta Bioquímica Clínica Latinoamericana (5-11) o en sus Suplementos (1)(12).
La sintomatología de las porfirias está asociada con la acumulación de los intermediarios del camino biosintético del hemo. El defecto enzimático en cada porfiria es hereditario, excepto en la Porfiria Cutánea Tardía (PCT) que también puede presentarse en una forma adquirida. Si bien todas las porfirias, excepto dos, se heredan en forma autosómica dominante, la penetrancia es variable y, en general, los portadores del gen afectado pueden permanecer asintomáticos durante toda su vida. La herencia de un alelo mutado disminuye la actividad de la enzima en aproximadamente un 50%; sin embargo, la cantidad de hemo sintetizado es suficiente para el normal funcionamiento del metabolismo celular. La presentación de la sintomatología clínica parecería entonces requerir de factores adicionales que afecten la biosíntesis del hemo, ya sea aumentando su demanda, produciendo una mayor disminución de la actividad enzimática o ambos factores a la vez (1). Los estudios bioquímicos, si bien son determinantes en pacientes sintomáticos (6), no son siempre absolutamente seguros para su correcto diagnóstico diferencial y menos aún en pacientes latentes en los cuales los parámetros bioquímicos están dentro de los valores normales. La detección e identificación de la mutación en el gen que codifica para la enzima afectada responsable de la porfiria, constituye en muchos casos, la única forma de identificar los portadores asintomáticos (8).
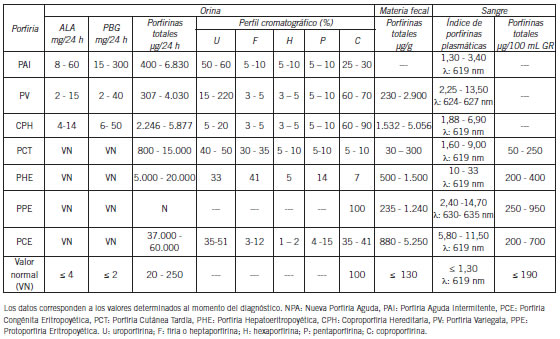
Las porfirias se pueden clasificar en cutáneas, agudas o mixtas de acuerdo con sus principales manifestaciones clínicas (Tabla I). Las porfirias agudas se caracterizan por la presencia de severos ataques con sintomatología abdominal aguda y un síndrome neurológico psíquico con ataques neurológicos intermitentes; en particular, los síntomas más comunes son dolores abdominales y gastrointestinales, hipertensión, taquicardia y diferentes manifestaciones que involucran disfunciones del sistema nervioso central (SNC), periférico y autónomo, como consecuencia de la acumulación de precursores, especialmente de ALA que se comporta como una neurotoxina. Las porfirias cutáneas presentan una manifestación cutánea en zonas expuestas debida a la formación de especies reactivas de oxígeno (ROS) por acción de la luz sobre las porfirinas acumuladas, cuya severidad depende del tipo de porfirina en exceso. Se las puede clasificar también en hepáticas, eritropoyéticas y hepatoeritropoyéticas, según el órgano principal de manifestación de la deficiencia enzimática (1). En la Tabla II se resumen los datos bioquímicos característicos de los distintos tipos de porfirias.
Tabla I. Aspectos generales de las porfirias.

Tabla II. Parámetros bioquímicos determinados en las diferentes porfirias.
Porfirias Agudas
PORFIRIA AGUDA INTERMITENTE
La Porfiria Aguda Intermitente (PAI) es la más común de las porfirias agudas y la primera en frecuencia en la población argentina (1:150.000). Surge como consecuencia de un defecto en la PBG-D, tercera enzima del camino biosintético del hemo. Se hereda con un carácter autosómico dominante y la expresión clínica ocurre después de la pubertad, aunque hay casos descriptos de PAI infantil (13)(14). A nivel bioquímico, los individuos sintomáticos presentan niveles aumentados de ALA y PBG en orina, responsables de los signos agudos (1).
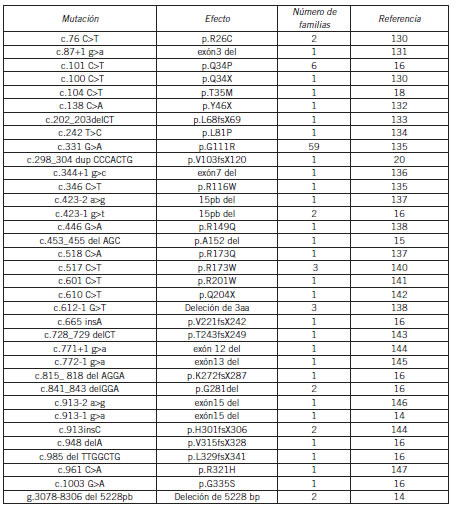
Hasta la fecha se identificaron más de 400 mutaciones diferentes en el gen de la PBG-D (Human Genome Mutation Database http://www.hgmd.org) que muestran la heterogeneidad molecular de la PAI. En el CIPYP se han estudiado hasta el momento 109 familias a nivel molecular; de 178 diagnosticadas bioquímicamente, se han detectado 22 mutaciones aún no descriptas en la literatura. El 54% de las familias PAI no relacionadas que fueron analizadas poseen la misma mutación (p.G111R) sugiriendo que tendrían un ancestro común (14-21) (Tabla III).
Tabla III. Mutaciones en el gen PBGD descriptas en la población argentina con PAI
PORFIRIA VARIEGATA
ácter autosómico dominante con penetrancia incompleta, y surge como consecuencia de una deficiencia parcial en la protoporfirinógeno oxidasa (PPox), la séptima enzima del camino metabólico del hemo. Además de presentar la sintomatología aguda, un alto porcentaje de los portadores de esta enfermedad pueden manifestar fragilidad cutánea y fotosensibilidad; es por esta variabilidad en los signos que también se la llama porfiria mixta. A nivel bioquímico, los individuos sintomáticos presentan niveles aumentados de ALA y PBG en orina durante el ataque; se observa además un aumento de porfirinas, principalmente PPIX que se excreta por heces y, en menor proporción, coproporfirina, responsables de la sintomatología cutánea (1).
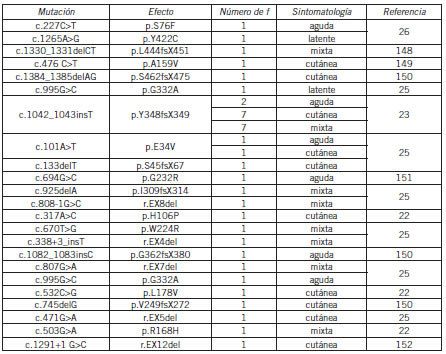
Hasta la fecha se han identificado alrededor de 170 mutaciones diferentes en el gen de la PPox (Human Genome Mutation Database http://www.hgmd.org) que causan PV. La frecuencia en la población argentina es de 1:500.000. En el CIPYP se diagnosticaron bioquímicamente, hasta el momento, 68 familias de las cuales 39 ya se han estudiado genéticamente. Se detectaron y caracterizaron 21 mutaciones aún no descriptas en la literatura (Tabla IV). Es notable que la mutación c.1043InsT se detectó en 16 familias argentinas aparentemente no relacionadas, siendo así la mutación más frecuente en esta población (41%); se ha encontrado que los portadores de esta mutación comparten un alelo, en base a un análisis de microsatélites, indicando un efecto fundador para la misma (22-28).
Tabla IV. Mutaciones en el gen PPOX descriptas en la población argentina con PV.
COPROPORFIRIA HEREDITARIA
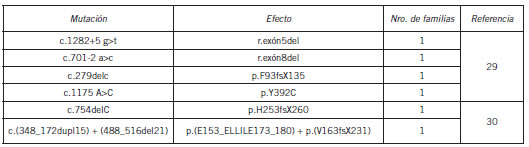
La Coproporfiria Hereditaria (CPH) se produce por una deficiencia en la coproporfirinógeno III oxidasa (CPGasa), sexta enzima del camino metabólico del hemo. La CPH es también una porfiria mixta; los pacientes pueden presentar síntomas abdominales y/o cutáneos. Bioquímicamente, la actividad disminuída de la CPGasa produce la acumulación hepática de coproporfirina y su excreción aumentada por orina y heces, mientras que durante los ataques, el PBG y el ALA urinarios también están incrementados. La CPH es muy poco común, incluso entre las porfirias. En Argentina se han diagnosticado sólo 17 familias. Hasta hoy, se han caracterizado 64 mutaciones en el gen CPO (Human Genome Mutation Database http://www.hgmd.org) y en las 6 familias argentinas estudiadas genéticamente se detectaron 6 mutaciones diferentes descriptas por primera vez en la literatura por el CIPYP (Tabla V) (29)(30).
Tabla V. Mutaciones en el gen CPO descriptas en la población argentina con CPH.
NUEVA PORFIRIA AGUDA
La Nueva Porfiria Aguda (NPA) es una enfermedad extremadamente rara, ya que hasta el momento se han publicado menos de 10 casos a nivel mundial, la mayoría de ellos varones jóvenes (31). Esta porfiria se caracteriza por una deficiencia severa en la enzima ALA-D y se presenta con los mismos síntomas que las otras Porfirias agudas; no se observan manifestaciones cutáneas. Bioquímicamente se puede diferenciar de la PAI en función de la excreción de PBG, que en el caso de la NPA se mantiene en niveles normales. La caída en la actividad enzimática de ALA-D es muy marcada, tanto en eritrocitos como en otros tejidos, con valores menores al 2% de actividad residual, produciendo una importante acumulación de ALA (32).
Entre los casos descriptos a nivel mundial se han indicado 12 alelos mutados (Human Genome Mutation Database http://www.hgmd.org). Al ser una enfermedad de herencia autosómica recesiva, el probando debe heredar un alelo mutado de cada padre y aunque estos no son sintomáticos, los valores de actividad enzimática están alrededor del 50% respecto al valor normal (32).
Porfirias Cutáneas
PORFIRIA CUTÁNEA TARDÍA
Una deficiencia en la uroporfirinógeno decarboxilasa (URO-D) es responsable de dos formas genéticas de porfiria cutánea: Porfiria Cutánea Tardía familiar o hereditaria (PCT-F) y Porfiria Hepatoeritropoyética (PHE). La primera se transmite en forma autosómica dominante, se caracteriza por la fotosensibilidad cutánea, frecuentemente asociada a una disfunción hepática y se precipita por factores ecogenéticos que incluyen la sobrecarga de hierro, compuestos policlorados, abuso de alcohol, hormonas, virus hepatotrópicos etc. La PHE se transmite con carácter recesivo, es más severa que la PCT-F y se la puede considerar como una forma homocigota de la PCT. En Argentina se ha encontrado solo un caso de PHE que porta mutaciones ya descriptas en PCT (33). También existe una forma adquirida de la PCT (PCT-A) con una actividad disminuída de URO-D sólo en hígado, que representa alrededor del 80% de la población PCT (1). Es la porfiria cutánea más común y tiene una frecuencia de 1:20.000 en Argentina.
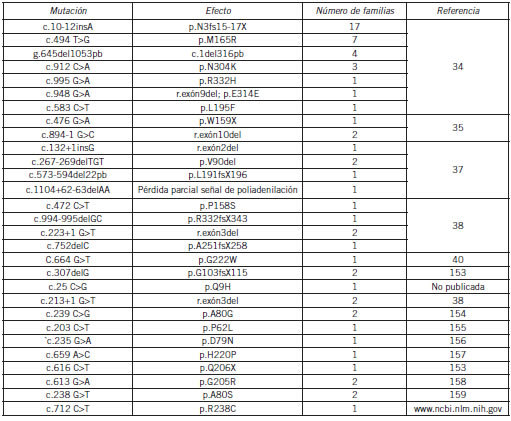
Hasta el momento se han descripto alrededor de 120 mutaciones diferentes en el gen de la URO-D (Human Genome Mutation Database http://www.hgmd.org). En 64 familias PCT-F argentinas no relacionadas se detectaron 19 mutaciones nuevas en el gen que codifica para la URO-D y dos mutaciones missense que ya habían sido reportadas en estado homocigota en familias con PHE. En 17 familias se detectó la mutación c.10-12insA, así, la prevalencia de esta mutación en la población PCT-F argentina es del 26,6%, siendo la más frecuente en este país. La segunda en prevalencia es la mutación p.M165R, entre ambas representan el 40% de los alelos mutados caracterizados hasta la fecha (Tabla VI) (34-40).
Tabla VI. Mutaciones en el gen UROD descriptas en la población argentina con PCT.
Si bien en general es una porfiria que se desencadena en la edad adulta, a la fecha estos autores han descripto 28 casos de PCT infantil, hasta ahora el mayor número hallado en la literatura (13).
PROTOPORFIRIA ERITROPOYÉTICA
La protoporfiria eritropoyética (PPE) se produce en el 95% de los casos por una pérdida de función de la enzima FECH, última etapa del camino biosintético del hemo, responsable de la inserción del hierro (Fe2+) en la molécula de PPIX.
La acumulación de PPIX en médula ósea, glóbulos rojos, plasma e hígado resulta en un incremento de la excreción biliar de este compuesto. Clínicamente, la PPE se caracteriza por una fotosensibilidad temprana, desde la niñez, debido al daño que causa en la piel la fotoactivación de la PPIX, con un severo ardor, que incluye hinchazón y eritema en áreas expuestas a la luz solar. En un 5-10% de los pacientes con PPE, la enfermedad crónica hepática puede llegar a ser una complicación importante siendo necesario, en ocasiones, el trasplante del órgano. El 95-99% de los pacientes PPE presentan una deficiencia parcial de la FECH con herencia autosómica dominante y penetrancia incompleta; se han descripto sólo 26 casos de herencia autosómica recesiva (1)(41).
En la actualidad se han detectado más de 190 mutaciones y 538 variantes polimórficas (Human Genome Mutation Database http://www.hgmd.org). Funcionalmente, estos polimorfismos pueden modular en trans la penetrancia de la enfermedad (42).
En la forma dominante, el fenotipo es el resultado de la herencia combinada de un alelo mutado y uno de baja expresión que produce el 75% de la cantidad normal de ARNm de la enzima (43)(44).
Se ha demostrado que mutaciones en el gen del ALA-S 2 que producen ganancia de función causan un tipo de protoporfiria dominante ligada al cromosoma X (PPDLX), que es responsable del 2% de los pacientes que manifiestan PPE (45).
La forma PPDLX de la enfermedad, a diferencia de la PPE producida por una falla en el gen FECH, presenta un grado de penetrancia cercano al 100%. En este caso se incrementa la tasa de formación de ALA que se metaboliza casi en su totalidad produciendo la acumulación de PPIX en glóbulos rojos que puede convertirse en hemo libre y ser exportado al citoplasma. Además, la acumulación de Zn-PPIX observada en PPDLX, indica que la FECH utiliza Zn como sustrato alternativo. La elevada producción de PPIX que supera la cantidad requerida para la hemoglobinización en células eritroideas es suficiente para producir fotosensibilidad y daño hepático a pesar de la actividad normal de la FECH.
En Argentina, la frecuencia de la PPE es de 1:900.000. De las 44 familias con PPE, 16 se han estudiado molecularmente, detectándose 15 mutaciones, 7 de las cuales son nuevas en la literatura (41)(46) (Tabla VII).
Tabla VII. Mutaciones descriptas en el gen FECH en la población argentina.

PORFIRIA CONGÉNITA ERITROPOYÉTICA
La Porfiria Congénita Eritropoyética (PCE) es una enfermedad autosómica recesiva de la cual sólo se han descripto en la literatura alrededor de 200 casos, aunque fue la primera porfiria detectada en 1889 en Escocia en 2 hermanos (47). Se produce por una disminución en la actividad de la uroporfirinógeno III sintetasa (URO-S), que cataliza la formación del URO III, el isómero fisiológico en la biosíntesis del hemo. Se acumulan grandes cantidades de los isómeros de la serie I, no fisiológica, que se oxidan a URO I y COPRO I provocando la lisis de los glóbulos rojos y la liberación de grandes cantidades de porfirinas que se excretan por orina y materia fecal. La URO I acumulada en plasma es la principal responsable de las manifestaciones clínicas: lesiones cutáneas severas en zonas expuestas que pueden llevar a la mutilación de los tejidos, hipertricosis, alopecia, eritrodoncia, síntomas que generalmente se presentan desde el nacimiento, aunque ya se han descripto 13 casos de manifestación tardía (11).
Se han encontrado en la literatura alrededor de 49 mutaciones responsables de PCE. En este laboratorio se han estudiado 4 casos infantiles y uno de manifestación tardía. No fue posible estudiarlos a nivel molecular a tres de los casos infantiles; en cuanto al cuarto caso, fue sometido a trasplante de médula ósea a los 6 años; la donante fue su hermana y hasta el momento que ha cumplido 19 años, se encuentra en buen estado de salud; porta una mutación missense (p.C73R) y una mutación de splicing en el otro alelo (c.627ins27pb). Curiosamente, en el caso de manifestación tardía, fallecido en la actualidad por causas ajenas a la porfiria, se encontró sólo una mutación, la p.C73R, pero sufría también de talasemia menor (11)(48).
Porfiria experimental
El análisis de los mecanismos de la deficiencia de hemo, y su impacto sobre las hemoproteínas vitales y la homeostasis tisular, es un tema de enorme importancia en la patogénesis y el tratamiento de muchos desórdenes.
Las drogas son los desencadenantes más comunes de las porfirias. Entre otros factores que pueden provocar una crisis se encuentran el ayuno, la dieta, infecciones o intoxicaciones diversas.
Se definen como porfirias experimentales las provocadas por xenobióticos en animales o cultivos de células aisladas, con el fin de conocer la etiopatogenia, los mecanismos alternativos tisulares, la fisiopatología, los rasgos clínico-bioquímicos y la posible prevención y terapéutica de las porfirias.
Los efectos producidos por los compuestos porfirinogénicos se observan tanto en individuos con porfiria como en individuos normales, aunque en estos últimos la inducción de una manifestación porfírica requiere la administración de una dosis mucho más elevada. Este fenómeno de inducción por drogas también puede observarse en animales de experimentación, así como en determinados tipos de cultivos de tejidos (17).
Todos los compuestos porfirinogénicos tienen en común la propiedad de estimular marcadamente la actividad del ALA-S hepática y, además, de disminuir el contenido del hemo hepático, ya sea por inhibición de su síntesis o por aceleración de su degradación (1). Se conoce una extensa lista de estas drogas porfirinogénicas, que son capaces de provocar una porfiria aún en ausencia de una predisposición genética, produciendo anormalidades metabólicas similares a las observadas durante el ataque agudo de las porfirias hereditarias humanas (/sites.google.com/site//cipypconicet).
En lo que se refiere a la porfiria experimental, la acción de drogas porfirinogénicas y en particular los anestésicos volátiles, este laboratorio ha sido pionero y ha publicado numerosos trabajos que han permitido incluir ciertos anestésicos en la lista de drogas prohibidas para su uso en individuos con porfiria. En las Figuras 3, 4 y 5 se muestra la multiplicidad de metabolismos afectados cuando dichos xenobióticos o el ALA mismo, fueron administrados a ratones control y modelos genéticos de porfirias agudas y no agudas.
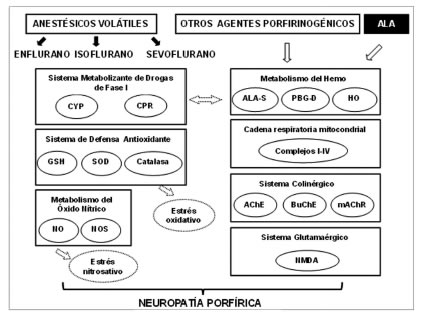
Figura 3. Efecto de los anestésicos volátiles, otras drogas porfirinogénicas y el ALA sobre diferentes metabolismos en ratones control.
ALA: Ácido d-aminolevúlico, ALA-S: Ácido d-aminolevúlico sintetasa, PBG-D: Porfobilinogeno deaminasa, HO: Hemo oxigenasa, CYP: Citocromo P-450, CPR: NADPH-citocromo P-450 reductasa, GSH: glutatión reducido, SOD: Superóxido
dismutasa, NO: Óxido Nítrico, NOS; Óxido Nítrico Sintasa, AChE: Acetilcolinesterasa, BuChE: Butirilcolinesterasa, mAChR: receptor muscarínico, NMDA: receptor nicotínico.
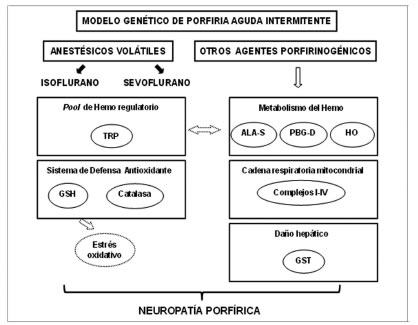
Figura 4. Efecto de los anestésicos volátiles sobre diferentes metabolismos en un modelo genético murino de PAI.
ALA-S: Ácido d-aminolevúlico sintetasa, PBG-D: Porfobilinógeno deaminasa, HO: Hemo oxigenasa, GSH: Glutatión reducido, TRP: Triptofano pirrolasa, GST: Glutation S-transferasa,
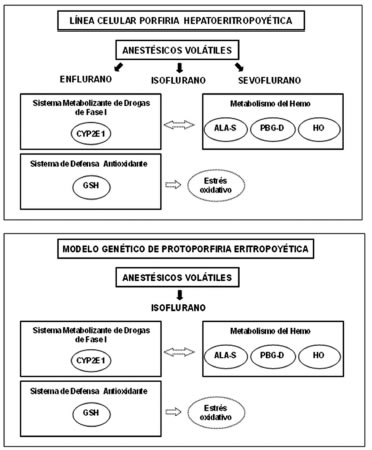
Figura 5. Efecto de los anestésicos volátiles sobre diferentes metabolismos en modelos de Porfirias no agudas.
ALA-S: Ácido d-aminolevúlico sintetasa, PBG-D: Porfobilinógeno deaminasa, HO: Hemo oxigenasa, CYP: Citocromo P-450, GSH: glutatión reducido.
PORFIRIA EXPERIMENTAL INDUCIDA POR ALA
Todas las porfirias hepáticas agudas presentan síntomas neurológicos cuya patogénesis permanece hasta ahora sin esclarecer (1)(49-52). Existen dos hipótesis principales para explicar el mecanismo de acción de las drogas porfirinogénicas que cuentan con cierto grado de evidencia experimental, la deficiencia de hemo y la toxicidad inducida por ALA (53). La deficiencia de hemo llevaría a la falta de hemoproteínas vitales como los citocromos respiratorios, con el consecuente daño del tejido nervioso.
Con respecto al ALA, se ha demostrado la capacidad del cerebro para generar su propio ALA; la síntesis probablemente ocurra en las células gliales desde las cuales es transferido a las neuronas. Estudios in vitro han revelado que el ALA tiene efectos sobre diversas funciones nerviosas; sin embargo, se ha puesto en duda su acción farmacológica a las concentraciones presentes en el tejido nervioso durante un ataque agudo de porfiria (54) (55). Por otro lado, se ha demostrado que el ALA puede acumularse y concentrarse en la corteza cerebral y que las ROS producidas por este compuesto pueden dañar diversos componentes celulares (56-58). Los efectos del ALA en el SNC podrían deberse a su unión a los receptores GABA (59). Además, se ha descripto que la inhibición de la captación de glutamato por el ALA podría ser la causa de las convulsiones inducidas por este precursor de la síntesis de hemo (60).
Los resultados de estos autores (Figura 3) han demostrado que el ALA puede acumularse en el SNC después de su administración, tanto aguda como crónica, afectando varios metabolismos como el sistema colinérgico y el metabolismo del óxido nítrico. En particular, el aumento de HO podría ser consecuencia del estrés oxidativo generado por la presencia del ALA, en concordancia con las alteraciones en el sistema de defensa antioxidante (61).
PORFIRIA EXPERIMENTAL INDUCIDA POR ANESTÉSICOS VOLÁTILES
El enflurano (2-Cl-1,1,2-trifluoroetildifluorometil-éter), el isoflurano (1-cloro-2,2,2-trifluoroetil difluorometil éter) y el sevoflurano (fluorometil 2,2,2-trifluoro-1 [trifluorometil]etil éter) son anestésicos volátiles muy utilizados para producir anestesia general (62)(63). La toxicidad hepática y renal de los anestésicos volátiles éteres fluorados se produce por su biotransformación, mediada por el citocromo P450 (CYP), a metabolitos tóxicos; el CYP2E1 es la isoforma responsable del metabolismo de estos compuestos en humanos (64).
ANESTÉSICOS Y METABOLISMO DEL HEMO EN RATONES CONTROL
La administración de los anestésicos enflurano, isoflurano y sevoflurano a ratones cepa CF1 modificó la actividad de algunas enzimas del metabolismo del hemo, reproduciendo algunos de los signos bioquímicos de la PAI (Figura 3) (65-69), efecto dependiente de la cepa y el sexo (70). Se produjeron alteraciones similares por la anestesia en animales bajo diferentes estados metabólicos: en animales alcoholizados (71), bajo inducción o depleción del CYP (9)(72), depleción de glutatión (73) e inducción de la enzima HO (74). Estos trabajos indicaron la potencial porfirinogenicidad de los anestésicos y las precauciones necesarias para su uso en pacientes con porfirias agudas.
ANESTÉSICOS Y METABOLISMO DEL HEMO EN MODELOS GENÉTICOS DE PAI
Teniendo en cuenta la multiplicidad de metabolismos afectados por las drogas porfirinogénicas o el ALA, razón por la cual es difícil establecer las causas de las manifestaciones neurológicas de esta enfermedad, y con el fin de incrementar el conocimiento sobre los mecanismos de acción de estas drogas en cerebro, se utilizaron ratones cepa C57BL6 knockout deficientes en la enzima PBG-D como modelo de PAI: T1 (homocigotas, actividad 55% reducida) y PAI (doble heterocigotas, actividad 70% reducida) (75) (Figura 4).
Se observaron alteraciones bioquímicas que dependieron del sexo y la mutación. Así, el isoflurano afectó aún más la actividad de la PBG-D, disminuida como consecuencia de la mutación en los animales homocigotas mientras que el sevoflurano causó un efecto similar pero en los ratones hembra, en los cuales, además, se produjo una importante inducción en la actividad de ALA-S (76). En los ratones PAI doble heterocigotas no se produjeron variaciones (Figura 4). En general, los efectos fueron más pronunciados que los observados en ratones cepa CF1, indicando una mayor sensibilidad del modelo a estos xenobióticos.
ANESTÉSICOS Y METABOLISMO DEL HEMO EN MODELOS GENÉTICOS DE PORFIRIAS NO AGUDAS
Si bien los anestésicos están principalmente relacionados con el desencadenamiento del ataque en las porfirias agudas, su administración en modelos genéticos de PHE (línea celular de linfocitos B) (77) y PPE (Fech m1pas/Fech m1pas) (78) causó alteraciones bioquímicas en la biosíntesis del hemo compatibles con las observadas en una porfiria aguda (Figura 5). Tanto el isoflurano como el sevoflurano indujeron la actividad del ALA-S y redujeron la actividad de la PBG-D cuando se agregaron a la línea celular de linfocitos B, mientras que no se observaron diferencias debido al tratamiento con enflurano. En el modelo de PPE la actividad de ALA-S hepática se indujo sin que se produzcan variaciones en la actividad de la PBG-D o de la HO.
Los resultados indicaron que el uso de estos anestésicos no sólo debe evitarse en el caso de porfirias agudas sino que también deben utilizarse con precaución en pacientes con porfirias hepáticas no agudas. Además, debe señalarse que el isoflurano produjo alteraciones en el metabolismo del hemo causando una reducción en la actividad de PBG-D y un aumento en la actividad de HO, cuando se administró a un modelo murino de hemocromatosis (Hfe-/-), enfermedad relacionada a alteraciones en la homeostasis del hierro y que frecuentemente está asociada a la PCT (79).
ANESTÉSICOS Y OTRAS DROGAS PORFIRINOGÉNICAS SOBRE METABOLISMOS INTERRELACIONADOS CON LA BIOSÍNTESIS DEL HEMO
Dentro de los estudios tendientes a elucidar los mecanismos que conducen a la neuropatía porfírica, se demostró que algunas drogas porfirinogénicas alteraban los sistemas colinérgico (80) y glutamaérgico (81), el sistema de defensa antioxidante (82), el sistema metabolizante de drogas (83)(84), el metabolismo del óxido nítrico (NO) (85)(86) y la cadena respiratoria mitocondrial (87) en encéfalo de ratones cepa CF1.
Las alteraciones descriptas podrían deberse a la acción directa de las drogas o sus metabolitos en cerebro, o al efecto del ALA sintetizado en el hígado y transportado al cerebro. El aumento de HO producido por los agentes porfirinogénicos estudiados, implica el disparo de mecanismos antioxidantes frente al estado de estrés oxidativo en el cerebro, lo que contribuiría y podría ser uno de los factores que conducen a la neuropatía porfírica. Estos resultados indicarían la multiplicidad de metabolismos afectados por las drogas porfirinogénicas o el ALA, razón por la cual es difícil establecer las causas de las manifestaciones neurológicas de esta enfermedad.
Los cambios observados en el SNC difieren de los producidos en el hígado para los mismos agentes porfirinogénicos. Estas diferencias sugieren que las alteraciones neurológicas observadas en la porfirias no se podrían explicar por simple extrapolación al SNC de los efectos deletéreos observados en el tejido hepático. De hecho, como ya se ha mencionado, no se conocen los mecanismos por los cuales se controla la formación de hemo en cerebro. Se ha demostrado que el ALA-S en cerebro de rata se induce por disminución del pool de hemo celular, pero no por la acción de agentes porfirinogénicos que actúan a través del CYP hepático. Por lo tanto, es necesario plantear nuevas hipótesis que contemplen la acción directa de los agentes porfirinogénicos, los cuales serían específicos para el SNC, o efectos secundarios debidos a productos originados en otros tejidos como consecuencia de la acción de dichos agentes.
Terapia fotodinámica
Las propiedades fotodinámicas de las porfirinas, que, como se ha visto, son causantes de la fotosensibilización cutánea en las porfirias, constituyen por otro lado, la base fundamental del procedimiento terapéutico conocido como terapia fotodinámica (TFD). Las porfirinas al ser irradiadas con luz de longitud de onda adecuada, pasan de su estado energético basal a un nivel excitado; esa energía es transferida a otras moléculas generando especies citotóxicas, principalmente oxígeno singulete, que producen la muerte celular. Esta propiedad se aplica para el tratamiento de tumores oncológicos y también en patologías no oncológicas, como la inactivación de microorganismos (bacterias, virus, parásitos) (Figura 6).

Figura 6. Mecanismo de acción de la Terapia Fotodinámica.
FS: Fotosensibilizante.
En el área de la terapia fotodinámica utilizando ALA (TFD-ALA), este laboratorio ha sido pionero. Ya en 1975, se describió la destrucción fotodinámica de callos de soya, tejido considerado como un tumor vegetal dada su alta división celular, cuando se agregaba ALA al medio de cultivo y se iluminaba (88). La base racional para la aplicación de TFD-ALA es la acumulación selectiva de PPIX inducida por ALA en los tumores, debido a una actividad reducida de la FECH y a una actividad relativamente aumentada de la PBG-D en las células malignas. Una acotación importante es destacar que la administración de ALA en ratones portadores de tumor, al menos en las dosis utilizadas en la TFD-ALA, no es citotóxica (89). Con respecto a la TFD-ALA, más de 75 artículos en revistas internacionales, 3 libros, capítulos en varios libros de TFD, numerosas conferencias, un Congreso Internacional sobre Terapia Fotodinámica del cáncer, realizado en Argentina, varios premios de organizaciones relacionadas con el cáncer, cientos de presentaciones en Congresos internacionales y nacionales, que se han publicado avalan la experiencia de estos autores en el tema (90)(91).
Más recientemente, se comenzaron a estudiar los mecanismos de muerte fotodinámica en células tumorales humanas y murinas, observando que dependen del tipo celular y del fotosensibilizante (FS) empleado (92)(93), pero no están dilucidados aún los efectos de la TFD sobre los mecanismos de señalización celular.
TFD-ALA EN CÉLULAS LEUCÉMICAS
En enfermedades como leucemias y linfomas, el empleo de transplante hematopoyético autólogo presenta el problema de la contaminación con células tumorales, las que luego de la re-infusión contribuyen a la recaída de la enfermedad. Es deseable entonces erradicar la fracción de células cancerosas sin causar daño a las células normales. Un posible método para este propósito es el purgado basado en tratamientos fotodinámicos (94)(95). Otra de las dificultades es la resistencia a drogas que suele generarse en los tratamientos antineoplásicos; algunos trabajos han descripto que la TFD no presenta resistencia cruzada con la quimioterapia (96), por lo que sería sumamente beneficiosa su aplicación en células leucémicas resistentes a drogas de quimioterapia.
Buscando entonces erradicar células malignas mediante la fotoactivación de porfirinas endógenas sintetizadas a partir de su precursor ALA, se realizaron estudios con líneas celulares de leucemia murina sensibles a drogas antineoplásicas (LBR-), y dos líneas derivadas, una resistente a doxorrubicina (LBR-D) y otra resistente a vincristina (LBR-V).
Se midió la síntesis de porfirinas bajo diferentes condiciones de incubación (concentración de ALA y tiempos) y se vio que la línea LBR-V fue la que sintetizó mayor cantidad de porfirinas. Cuando se evaluó la viabilidad celular mediante el método del Tripan Blue luego del tratamiento lumínico utilizando un banco de dos tubos fluorescentes (18 J/cm2), se observó que la línea parental (LBR-) fue más sensible al tratamiento fotodinámico que las líneas resistentes LBR-D y LBR-V. Mediante microscopía confocal, utilizando marcadores mitocondriales y lisosomales, se vio un patrón similar de localización de PPIX intracelular en las tres líneas. Si bien inicialmente la PPIX se detectó principalmente en mitocondrias y parte en lisosomas, a las 20 h de incubación el patrón de fluorescencia se volvió más difuso, indicando una redistribución del fotosensibilizador en el citoplasma celular. Esta localización intracelular sugiere que la apoptosis podría constituir un mecanismo de muerte asociado a la TFD, por lo que se estudió la morfología mitocondrial mediante tinción con MitoTracker Green. En las tres líneas se observó una pérdida de la integridad mitocondrial a medida que aumentó la dosis lumínica. Se evaluó entonces la inducción de apoptosis utilizando naranja de acridina-bromuro de etidio. Se observó un incremento en la morfología apoptótica en función de la dosis lumínica y del tiempo post-TFD. La línea LBR-V160, que fue la que mayor cantidad de PPIX sintetizó, a 2 h post-TFD presentó el mayor porcentaje de apoptosis, indicando que sería la más sensible al tratamiento. En todas ellas la inducción de necrosis fue muy baja. Se evaluó la liberación de citocromo c al citoplasma utilizando un anticuerpo específico. Se produjo un aumento en la liberación del mismo cuando las células se trataron con la TFD, sugiriendo la activación de la vía apoptótica intrínseca. Se estudió también la activación de apoptosis lisosomal utilizando el inhibidor de catepsina D, pepstatina A (pA). En las líneas celulares resistentes se observó una disminución en los niveles de apoptosis al utilizar pA, en comparación con las células tratadas sólo con TFD, mientras que en la línea sensible LBR- no se encontraron diferencias.
Se observó, por lo tanto, que las tres líneas celulares respondieron a TFD-ALA, en concordancia con resultados obtenidos por otros autores, quienes describieron que la TFD pudo revertir la resistencia a drogas en distintos tipos celulares (97)(98).
Los resultados obtenidos sugieren entonces que la TFD-ALA constituye una alternativa eficaz para la erradicación de células malignas remanentes en el tratamiento de leucemias resistentes a quimioterapia, y contribuye a dilucidar los mecanismos de acción subyacentes a la TFD.
ESTRÉS OXIDATIVO INDUCIDO POR LA TFD-ALA.
Con el objeto de investigar los mecanismos intervinientes en la citotoxicidad de la TFD-ALA, se estudiaron las especies reactivas involucradas en el fotodaño luego de incubar las células con ALA seguida de una irradiación con luz blanca, y se evaluó la producción de radicales peroxilo, peróxido de hidrógeno, radicales hidroxilo, anión superóxido, y el daño mitocondrial determinado mediante la despolarización de la membrana mitocondrial. En todos los casos los parámetros de estrés oxidativo medidos aumentaron con el tiempo de irradiación en las tres líneas estudiadas, y fueron mayores en la línea sensible LBR- (99). Estos resultados indican la importancia de las mitocondrias en los efectos de la TFD.
TERAPIAS COMBINADAS EN LÍNEAS CELULARES LEUCÉMICAS: QUIMIOTERAPIA Y TFD-ALA.
Como se indicara anteriormente, el transplante de médula ósea asociado a quimioterapia es una de las modalidades terapéuticas utilizadas en el tratamiento de la leucemia, siendo una limitación importante la presencia de células malignas remanentes y la aparición de fenotipos resistentes a multidrogas. Por otra parte, la aplicación de terapias combinadas con diferentes mecanismos de acción contribuye a incrementar la efectividad y minimizar las reacciones adversas en el tratamiento del cáncer. Resultó de interés entonces estudiar los efectos de la TFD-ALA combinada con DOX y VCR, en la línea celular de leucemia murina LBR- sensible a drogas de quimioterapia.
Se observó un efecto sinérgico al utilizar las terapias combinadas, demostrado por una disminución significativa de la supervivencia celular con respecto a cada terapia en forma independiente; este efecto fue mayor para el tratamiento con VCR en comparación con DOX, y la mortalidad fue superior a la obtenida con el doble de dosis de DOX y VCR (Tabla VIII).
Tabla VIII. Efecto del tratamiento con TFD, DOX o VCR, solos y combinados, sobre la supervivencia de las células LBR-D160.

Estos resultados indican que la combinación de TFD y quimioterapia puede constituir una alternativa terapéutica beneficiosa, generando la posibilidad de lograr una determinada eficacia disminuyendo las dosis de quimioterapia (en este caso, DOX y VCR en un 50%), lo que contribuiría a minimizar los efectos secundarios adversos de la misma y el desarrollo de fenotipos celulares resistentes a drogas (100). En base al efecto sinérgico obtenido al utilizar las terapias combinadas en un modelo in vitro, resultó interesante evaluar la respuesta de esta combinación en un modelo in vivo. Además, hasta el presente, en la bibliografía no se publicaron trabajos en los que se haya estudiado el efecto combinado de la TFD-ALA y drogas de quimioterapia en modelos in vivo de leucemia, aunque algunos autores estudiaron esta combinación empleando otros FSs y otros agentes antineoplásicos en distintos modelos de cáncer in vivo (101-105). Con el fin de evaluar los efectos de las terapias combinadas, ratones BALB-C se inocularon vía iv con células LBR- tratadas previamente en cultivo con ALA-TFD, DOX, VCR, o la combinación de tratamientos TFD+quimioterapia, dando lugar a los siguientes grupos experimentales: células LBR- no tratadas (grupo control), tratadas sólo con TFD, DOX, VCR, y las combinaciones TFD+DOX, TFD+VCR. Los animales se sacrificaron a los 30 días y se evaluó presencia y grado de infiltración tumoral mediante análisis histológico en timo, riñón, hígado, bazo, corazón, pulmón, ganglios linfáticos y cerebro. En el grupo inoculado con el vehículo no se observó infiltración, mientras que el inoculado con las células sin tratar se observó una gran intensidad de infiltración, en el 87,5% de los órganos. El grupo inoculado con células sometidas a TFD mostró un 50% de órganos infiltrados (principalmente ganglios linfáticos), pero con menor intensidad que el grupo sin tratar. En los animales inoculados con células tratadas con DOX o VCR se observaron células tumorales en el 25% y el 75% de los órganos, respectivamente. Utilizando la combinación DOX+TFD o VCR+TFD, no se evidenció infiltración de células leucémicas en ninguno de los órganos estudiados (Tabla IX) (106).
Tabla IX. Porcentaje de órganos afectados con infiltración leucémica (IL) para los distintos grupos de tratamiento.
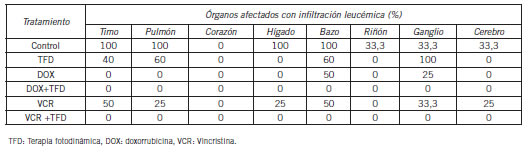
El beneficio al utilizar TFD en combinación con DOX o con VCR en este modelo in vivo, concuerda con los resultados obtenidos previamente in vitro. Canti et al (107) utilizaron combinaciones similares en modelos in vivo de leucemia, hallando resultados sinérgicos, e incluso aditivos aplicando TFD con ftalocianina de alumino como FS en combinación con DOX y con cisplatino, así como también combinando tres terapias en forma simultánea: TFD basada en ftalocianina, junto con linfocitos inmunes (inmunoterapia) y las drogas de quimioterapia vinorelbina o cisplatino.
Teniendo en cuenta los resultados previamente descriptos, tanto a nivel in vitro, como in vivo, queda en evidencia el beneficio de la utilización de TFD-ALA en combinación con drogas de quimioterapia como un método eficaz para erradicar las células malignas durante el purgado de médula ósea en pacientes con leucemia, sugiriendo una potencial alternativa terapéutica que permitiría disminuir las dosis de quimioterapia, minimizando así sus limitaciones y efectos adversos.
TFD-ALA EN CÉLULAS DE CÁNCER DE PULMÓN
El cáncer de pulmón de células no pequeñas (NSCLC) es la histología más frecuente (80%) de este tipo de tumores. Actualmente, se aplica TFD a tumores de pulmón obstructivos y microinvasivos. Luego de administrar el FS que se acumula selectivamente en el tejido tumoral, se fotoactiva por vía broncoscópica con luz de longitud de onda adecuada, y se generan ROS que provocan daños celulares irreversibles llevando a la muerte celular.
Se realizaron estudios con células de adenocarcinoma de pulmón humano A549 (NSCLC) utilizando el modelo de crecimiento en cultivos bi y tridimensionales.
TFD-ALA EN CULTIVOS BI Y TRIDIMENSIONALES DE CÉLULAS DE ADENOCARCINOMA DE PULMÓN
El cultivo de células en esferoides multicelulares (EMCs) representa un modelo in vitro tridimensional (3D) de tumores sólidos, más representativo de lo que ocurre in vivo respecto del cultivo en monocapa. Es útil para el estudio de distintas terapias antitumorales, y apropiado para el estudio de muerte celular post-TFD, dado que simula las condiciones de microtumores independientemente de la vasculatura, cuyo rol en la erradicación de tumores por TFD es controversial. Se realizó la puesta a punto de las condiciones adecuadas para el cultivo de esferoides en la línea A549 y se comparó la respuesta de síntesis de porfirinas y viabilidad post-TFD en las conformaciones 2D y 3D del cultivo. Sembrando 5x104 células/mL sobre agar 3%-RPMI (1:1) se obtuvieron esferoides compactos de 100-700 μm a los 7 días.
En el cultivo en esferoides se llegó a un plateau en la síntesis de porfirinas a una concentración de ALA mayor que en el cultivo en monocapa. A plazos cortos (3 h) se observó que el aumento de la síntesis de porfirinas en función del tiempo de incubación, fue similar para ambos tipos de cultivo, pero a tiempos largos (16 h) la cinética de síntesis en la monocapa fue más rápida que en los esferoides. En el modelo 3D, por microscopía confocal, se observó una distribución de porfirinas uniforme en los distintos planos del esferoide.
Se analizaron, tanto en la monocapa como en los esferoides la viabilidad celular por el método de MTT y la muerte celular por tinción con naranja de acridina y bromuro de etidio, luego de la incubación con ALA y posterior irradiación. Las curvas de supervivencia post- TFD-ALA en esferoides mostraron un ligero corrimiento (2 min) de la dosis lumínica que produce un 50% de muerte celular (DL50) y una mayor supervivencia de células en esferoides (31,9±8,7%) respecto de la monocapa (9,3±0,8%) para una irradiación de 20 min. Estos resultados permiten inferir que, si bien la síntesis de porfirinas en los esferoides es del mismo orden que en la monocapa, la accesibilidad de la luz a las células del interior del esferoide es un factor limitante para la efectividad de la terapia en este modelo de conformación 3D propia de los tumores (108).
ALTERACIONES CELULARES EN UN MODELO TRIDIMENSIONAL DE CÉLULAS DE ADENOCARCINOMA DE PULMÓN HUMANO LUEGO DE LA APLICACIÓN DE ALA-TFD
En el modelo 3D, 1 h después de la irradiación, se visualizó el fotodaño mediante diferentes métodos: tinción con NA/BE, Hoechst y H/E. Se observó pérdida de compactación de la estructura del EMC con desprendimiento de células muertas de la superficie, disrupción de la membrana, aumento de la granularidad del citoplasma, gran cantidad de núcleos condensados y con tinciones atípicas. La tinción para mucopolisacáridos de secreción (PAS) indicó en EMCs no tratados fuerte marcación que se pierde luego de la TFD. La proliferación celular fue inmunodetectada con Ki67, mostró en los EMCs control, un gradiente de células proliferativas desde la periferia hacia el centro. Luego de la TFD las células positivas se restringen a la periferia en un cordón de células proliferativas con morfología relativamente conservada y escasa marca hacia el interior de la estructura.
La condición hipóxica basal en los EMCs no tratados mostró una expresión de HIF1α moderada y heterogénea de localización nuclear, que se incrementó con una dosis lumínica baja (5 min) y se perdió con 10 y 20 min de irradiación. Se explicaría así la menor generación de ROS comparada con la monocapa, los signos de apoptosis disminuidos (condensación nuclear, liberación de citocromo c, despolarización mitocondrial, activación de caspasa-3, Anexina) e indicios de necrosis y autofagia (expresión de Bcl-2 y LC3-II).
La detección de ROS no fue significativamente diferente en presencia y en ausencia de antioxidantes de alto grado de protección (ascorbato y trolox, según los cultivos en monocapa), pero por microscopía de fluorescencia se vio un efecto protector a nivel superficial en las capas externas de los EMCs (NA/BE), sugiriendo que el fotodaño a los tejidos normales puede ser atenuado mediante la administración de agentes antioxidantes (109).
Los resultados muestran que la estructura interna y la diferenciación de las células en el EMC se ven alteradas por la TFD-ALA, señalando que el efecto no es únicamente superficial, sino también en las capas internas.
CÉLULAS DE ADENOCARCINOMA DE PULMÓN HUMANO SENSIBLES Y RESISTENTES A TFD-ALA
La TFD se aplica en numerosas patologías tumorales, pero la aparición de células resistentes es un efecto no deseado. Para identificar qué cualidades confieren a las células la capacidad de sobrevivir a la TFD, se trataron células de adenocarcinoma de pulmón humano A549 con TFD-ALA. Se generó un linaje celular resistente a TFD-ALA luego de 12 ciclos sucesivos (C-12), que acumuló un 50% menos de porfirinas (control: 19,9±1,2 ng porfirinas/105 células; C-12: 9,5±0,7 ng porfirinas/105 células). La fotosensibilización con PPIX exógena indujo recuperación parcial de la sensibilidad a TFD en células C-12 (supervivencia irradiación 10 min: control ALA: 39,5%; control PPIX: 27,5%; C-12 ALA: 10,0%; C-12 PPIX: 75,0%). La menor sensibilidad de las C-12 a la PPIX-TFD no se debe a una menor acumulación de PPIX, pues es similar en ambos tipos celulares (77,1±2,2 ng porfirinas/105 células control; 80,6±4,8 ng porfirinas/105 células C-12). No hubo diferencias notables en la morfología (tinciones con azul de toluidina y Hoechst) ni en la capacidad de migración (ensayo de la herida). En cuanto a parámetros asociados a apoptosis: en las C-12 se detectó despolarización mitocondrial sin liberación de citocromo c, activación de caspasa-3 y se observó expresión de Bcl-2. Siendo NFκB un factor sensible al estado redox, es relevante en la supervivencia a TFD. Se analizó la expresión de IκB y la subunidad p65 de NFκB en las fracciones citosólicas y nucleares respectivamente, de células parentales y C-12, luego de la TFD. En células sensibles se detectó IκB; en las C-12, se detectó NFκB en todos los tratamientos, y un patrón de bandas de IkB similar al de células sensibles. Los resultados sugieren que la activación de NFκB es un evento relacionado con un efecto a largo plazo de la TFD. Las células que reciben un único tratamiento fotodinámico sólo activan esta respuesta con dosis subletales, menores a la DL50 (110).
EFECTO DE SECUESTRANTES DE ROS EN LA TFD-ALA
A fin de evidenciar la participación de las ROS en la inducción temprana de la muerte celular, se determinó el grado de protección de distintos agentes secuestrantes: glutation reducido, manitol, triptofano y ascorbato (Asc) en la TFD-ALA de células A549, LBR-, LBR-D160 y LBR-V160. Para estudiar en forma comparativa el efecto de los compuestos secuestrantes se determinó el grado de protección (GP) como la supervivencia post-TFD en presencia del compuesto relativa a la supervivencia en ausencia del mismo. Utilizando tiempos de iluminación entre 5 y 10 min, a las concentraciones estudiadas (0,5-10 mM), ninguno de los compuestos fue fotoactivo en ausencia de ALA. En células A549 el Asc mostró el mayor GP (% apoptosis TFD 7 min: 16,3±1,3% vs. 65,8±23,7%; % apoptosis TFD10 min: 48,3±2,7% vs. 98,4±3,5%; con y sin Asc respectivamente). Con el resto de los compuestos se observó efectividad a los 10 minutos de iluminación pero no a los 7 min. En las líneas leucémicas, Asc protegió a las tres líneas celulares, y el efecto fue mayor en las líneas resistentes cuando se irradiaron durante 10 min.
Los resultados obtenidos contribuyen a dilucidar los mecanismos de acción de la TFD, demostrando la participación de las ROS en el proceso de muerte celular, y proveen una herramienta para evaluar las condiciones a tener en cuenta para optimizar la eficacia de la TFD, ya que secuestrantes como trolox o Asc serían efectivos en la prevención clínica del fotodaño a tejidos no tumorales (111).
Microorganismos hemo-deficientes: Trypanosoma cruzi
Si de aspectos benéficos y perjudiciales de las porfirinas se trata, los microorganismos hemo-deficientes constituyen un ejemplo donde convergen ambos efectos. El carácter de hemo-deficientes se debe a que poseen un camino biosintético de las porfirinas total o parcialmente bloqueado; en este caso, el suministro de compuestos hémicos es necesario para asegurar la supervivencia de estos microorganismos. Pero por otro lado, cuando lo que se suministra e incorpora excede a lo requerido para el normal funcionamiento de la célula, entonces se manifiesta el conocido efecto citotóxico generado por las porfirinas acumuladas. Dentro de los organismos incapaces de sintetizar hemos. Se seleccionó a Trypanosoma cruzi, el agente causal de la enfermedad de Chagas, como objeto de estudio en este laboratorio.
Referido al camino biosintético de las porfirinas, en base a la determinación de precursores, porfirinas y actividades enzimáticas en epimastigotes de T. cruzi (112) (113) se concluye que el parásito conservaría funcionales las enzimas de localización mitocondrial, ALA-S y FECH (Figura 7). A continuación se detalla éste y otros estudios referidos a la vía metabólica del hemo en el parásito.
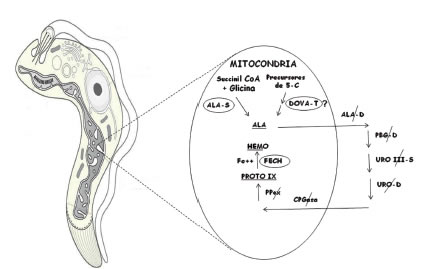
Figura 7. Camino biosintético de las porfirinas en Trypanosoma cruzi. Se esquematiza la localización intracelular y citosólica de la vía metabólica, remarcando las enzimas funcionales (ᴑ) y las que no mostraron actividad ( ∕ ).
ALA-S: Ácido d-aminolevúlico sintetasa, DOVA-T: Ácido 4,5-dioxovalérico transaminasa ALA-D: Ácido d-aminolevúlico dehidrasa, PBG-D: Porfobilinógeno deaminasa, UROgen III-S: Uroporfirinógeno III sintetasa, URO-D: Uroporfirinógeno decarboxilasa, CPGasa: Coproporfirinogenasa, PPox: Protoporfirinógeno oxidasa, FECH: Ferroquelatasa.
SÍNTESIS DE ALA Y HEMO EN T. cruzi
Empleando cultivos de epimastigotes de T. cruzi, cepa Tulahuen,Tul 2, se investigaron ambas vías de síntesis de ALA (114), la del ALA-S y la vía en la que a partir de precursores de 5 átomos de C se forma ALA manteniendo la integridad del esqueleto carbonado (115)(116), conocida como vía de 5-C (esta vía se encargaría de sintetizar ALA, principalmente en plantas y microorganismos fotosintéticos). Considerando al ácido 4,5-dioxovalérico (DOVA) como intermediario de la vía de 5-C, la enzima DOVA-transaminasa (DOVA-T) sería la encargada de catalizar la síntesis de ALA a partir de DOVA (117-119). A pesar de no detectar actividad de ALA-S in vitro, se midieron niveles significativos de ALA intracelular y gran cantidad de ALA excretado al medio. El 98% del ALA sintetizado por el parásito es excretado al medio extracelular; esta eliminación evitaría el efecto citotóxico en el interior de la célula y pone de manifiesto la falta de funcionalidad de la vía encargada de metabolizarlo (ALA-D y demás enzimas citosólicas). Referente a la actividad del ALA-S, en este laboratorio se detectó la presencia de una molécula no proteica, de bajo peso molecular, inestable al calor, que es capaz de inhibir la actividad de esta enzima proveniente de bacterias fotosintéticas (114). La capacidad inhibitoria de esta molécula se correlaciona con los niveles intracelulares de hemo. Respecto a la vía de 5-C, ni intra- ni extracelularmente se detectó DOVA y se midieron niveles muy bajos de actividad de DOVA-T, que serían insuficientes para justificar la cantidad de ALA sintetizado. Estos resultados llevaron a concluir que el parásito es capaz de sintetizar ALA a pesar de no poder convertirlo en porfirinas. La síntesis de ALA sería catalizada principalmente por la vía del ALA-S, cuya actividad estaría regulada por los niveles de hemo directa e indirectamente a través de la actividad del factor inhibitorio detectado en el parásito.
Teniendo en cuenta que ya se ha dilucidado la secuencia del genoma del parásito, se está estudiando la expresión de genes presentes en T. cruzi, que según un análisis bioinformático previo (GeneDB, Blast, ORF Finder) serían candidatos para codificar con alto score para una proteína con actividad de ALA-S. Es interesante esclarecer el significado fisiológico que tendría en el parásito, la presencia de una enzima involucrada específicamente en una vía que no es metabólicamente funcional.
En lo relacionado con la FECH, se detectó actividad en la fracción mitocondrial de epimastigotes de T. cruzi, cepas Tulahuen e Y (120). La enzima manifestó actividad in vitro tanto con PPIX como con deutero- y mesoporfirina como sustratos; en cuanto al metal a insertar en el anillo porfirínico, los mayores valores de actividad se obtuvieron con Fe2+ y Zn2+ (100 µM), a la misma concentración con Cu2+ y Co2+ no se observó actividad, mientras Mn2+ y Mg2+ se manifestaron como potentes inhibidores. Según el sustrato porfirínico utilizado, la enzima fue fuertemente inhibida (entre 40 a 75% de inhibición) por Tritón X-100, lo que manifiesta la conocida asociación a membrana requerida en la catálisis.
La FECH del parásito en presencia de PPIX y Fe2+ como sustratos mostró, a diferencia de la enzima de humanos, una fuerte inhibición con el agregado de Cu2+. Este hallazgo sería importante a tener en cuenta en una terapia que actuara sobre el parásito sin afectar al huésped. Actualmente está en estudio en este laboratorio, la identificación de una secuencia a nivel de genoma que codifique para la Hemo-S de T. cruzi.
EFECTO DE LOS COMPUESTOS HÉMICOS SOBRE T. cruzi
Trabajando con epimastigotes de T. cruzi, cepa Tulahuén, Tul 2 cultivados en medio líquido, se evaluó el efecto de distintas concentraciones de hemina (entre 0-30 mg/L) sobre la diferenciación celular, contenido total de proteínas, enzimas del sistema de defensa antioxidante del parásito, actividad y expresión de HO (121).
Concentraciones de hemina entre 5 y 10 mg/L resultaron ser las óptimas para obtener epimastigotes en cultivo, concentraciones mayores disminuían la velocidad de crecimiento y modificaban la morfología de la célula. Los cambios se correspondían con la transformación del estadío epimastigote a amastigote. Respecto al efecto de la hemina sobre la síntesis de proteínas, se sabe que altos niveles de hemina estimulan la transcripción al permitir la fosforilación del factor de iniciación eIF-2α (122). Investigando el efecto de la hemina sobre el contenido total de proteínas en parásitos se pudo observar que hasta alcanzar la concentración óptima, el comportamiento fue acorde a lo esperado, ya que a medida que aumentaba la concentración de hemina también aumentaba la cantidad de proteínas. Para niveles de hemina por encima del óptimo para el crecimiento, no sólo se invierte este comportamiento sino que además se produce degradación de las proteínas existentes. Se han descripto resultados similares en Leishmania donovani, donde para concentraciones de hemina del orden de 30 mg/L se observó una notable disminución en el contenido de proteínas en general y de β-tubulina en particular (123).
Asimismo, en células de mamíferos se ha observado una degradación de proteínas catalizada por hemina (124).
Conocida la capacidad de la hemina de generar ROS y modificar el estado de óxido reducción de la célula, se evaluó el estado del sistema de defensa antioxidante del parásito al variar los niveles de hemina en el medio de cultivo. Los Trypanosomátidos carecen de actividad de catalasa y glutation peroxidasa, su sistema antioxidante involucra a las enzimas superóxido dismutasa (SOD), ascorbato peroxidasa (APx), triparedoxin peroxidasa (TrxPx) y antioxidantes de bajo peso molecular, principalmente trypanotiona (N1,N8- bisglutationillespermidina (Try) (125). Otra enzima interviniente es la Trypanotiona reductasa (TryR), dependiente de NADPH que se encarga de regenerar la Try reducida, metabolito esencial para mantener la actividad de APx y TrxPx. La acción combinada de APx, TrxPx y TryR es de primordial importancia para mantener una concentración baja de H2O2. En la Figura 8 se esquematiza en forma comparativa el sistema de defensa antioxidante presente en el parásito (A) y en una célula de mamífero (B). Las diferencias existentes entre ambos sistemas, justifican la distinta toxicidad para el parásito y hospedador, que muestran las drogas antiparasíticas cuyo mecanismo de acción involucra procesos oxidativos o generación de especies oxidantes. Así, el sistema antioxidante del parásito constituye un blanco interesante al momento de diseñar drogas para combatir la enfermedad de Chagas.
ásito (actividades de SOD, APx y TryR y contenido de grupos tioles), se observó para concentraciones de hemina menores de 15 mg/L, que tanto las enzimas como el contenido de tioles aumentó progresivamente con la concentración de hemina, para luego decaer abruptamente a concentraciones mayores (hasta 30 mg/L). El daño oxidativo producido por altas concentraciones de hemina no sería contrarrestado por el sistema de defensa antioxidante y podría justificar la degradación de proteínas mencionada anteriormente. Para las mismas condiciones de cultivo se investigó la actividad y expresión de HO (121). Esta enzima no sólo degrada el hemo sino que ejerce un importante efecto protector contra el estrés oxidativo, debido al poder antioxidante adjudicado a la biliverdina y bilirrubina (4)(126). Tanto la actividad como expresión de la HO se incrementaron al aumentar la concentración de hemina, dicho incremento fue significamente mayor para la expresión (6-7 veces) que para la actividad (alrededor de 3 veces). Por análisis de Western blot se identificó que la isoforma evaluada se correspondía con la HO-1. En base a este comportamiento es evidente una correlación positiva entre actividad y expresión de la enzima, mostrando la expresión mayor sensibilidad frente a los cambios en los niveles de hemina.
Si se compara el comportamiento de las enzimas del sistema antioxidante con el de la HO, se ve que mientras las enzimas antioxidantes disminuyen su actividad, la HO se induce significativamente. Este comportamiento pone de manifiesto el diferente modo de acción y la complementación de ambos sistemas antioxidantes.
En base a estos resultados se concluyó que tanto la deprivación de hemo como su acumulación son perjudiciales para T. cruzi. Con el fin de utilizar estos resultados con fines terapéuticos, nuestro propósito es interferir en la captación y/o utilización del hemo, para producir, por sobrecarga o depleción, la muerte del parásito.
ACTIVIDAD ANTI-T. cruzi DE COMPUESTOS ANÁLOGOS AL HEMO
Teniendo en cuenta que la hemina y porfirinas relacionadas ejercen una importante acción citotóxica y la avidez del parásito por este tipo de compuestos, se investigó el efecto de la cianocobalamina (análogo estructural del hemo) in vitro sobre epi-, tripo- y amastigotes de T. cruzi e in vivo en un modelo murino agudo de la enfermedad de Chagas (127).
Estructuralmente la vitamina B12 (Figura 8) es una cobalamina que resulta de la unión asimétrica de cuatro anillos pirrólicos, formando un grupo macrocíclico casi planar (núcleo corrínico) en torno a un átomo central de cobalto. El anillo corrínico (derivado de la URO III) es parecido al anillo porfirínico y se diferencia de éste por el carácter asimétrico de las uniones entre los grupos pirrólicos, por lo tanto puede ser utilizado como análogo estructural de la hemina.
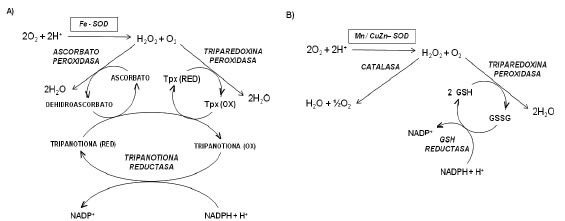
Figura 8. Sistema de defensa antioxidante en Tripanosomátidos (A) y en células de mamíferos (B).
Fe-SOD: superóxido dismutasa dependiente de Fe; Tpx: triparedoxina peroxidasa. MnSOD: superóxido dismutasa dependiente de Mn; Cu-Zn-SOD: superóxido dismutasa dependiente de Cu y Zn; GSH: glutatión reducido; GSSG: glutatión oxidado.
La vitamina B12 produjo in vitro una marcada actividad inhibitoria sobre los tres estadíos del parásito epi-, tripo- y amastigotes. Los valores de IC50 obtenidos (IC50 epi 2,42±0,54 µM; IC50 amas 10,69±1,50 µM y IC50 trip 9,46±1,20 µM) resultaron ser de 2 a tres veces menores a los del Benznidazol (Bnz) utilizado como droga de referencia. La vitamina B12 incrementó su actividad antiparasítica en presencia de un reductor como el ácido ascórbico. A pesar de que la cianocobalamina, al igual que la hemina, induce en el parásito muerte celular por daño oxidativo, el antioxidante potencia su efecto citotóxico debido a la conocida acción prooxidante que un reductor manifiesta en presencia de metales de transición, como ser Fe, Cu o Co (128)(129).
Al tratar de dilucidar el mecanismo de acción de la vitamina B12 empleando epimastigotes de T. cruzi se observó por citometría de flujo que la misma incrementa el estado oxidativo intracelular, genera anión superóxido, a la vez que aumenta la actividad de SOD (50%) y APx (sólo un 20%) sin modificar la actividad de TryR. En base a estos resultados se postuló que la vitamina B12 ejerce su acción a través de la secuencia de reacciones que se esquematiza en la Figura 9.

Figura 9. Estructura de la vitamina B12 y mecanismo propuesto para su actividad antiparasítica.
Ver descripción en el texto.
En primer lugar, por acción de un compuesto antioxidante (CAO) se produce la reducción del cobalto (presente como Co3+ en la vitamina B12) a Co2+ y luego el Co2+ reduce el O2 a anión superóxido, el anión superóxido a peróxido de hidrógeno y el peróxido de hidrógeno a radical hidroxilo. En conclusión, la vitamina B12 actuaría sobre el parásito como un generador de ROS y su administración, conjuntamente con un agente reductor, podría tenerse en cuenta para nuevas modalidades terapéuticas a emplear en el tratamiento de la enfermedad de Chagas.
Teniendo en cuenta estos resultados, empleando ratones infectados en forma aguda con T. cruzi, se evaluó, sobre diferentes lotes de animales, el efecto del tratamiento con B12, B12+ácido ascórbico (vitamina C), Bnz y B12+ácido ascórbico+Bnz. El tratamiento sólo con B12 fue capaz de reducir en aproximadamente un 50% la parasitemia, siendo esta disminución 2 veces mayor en presencia de ambas vitaminas. Mientras en el pico de la parasitemia el nivel de parásitos circulantes fue similar para los ratones tratados con las vitaminas, con Bnz, o con Bnz + ambas vitaminas se observaron diferencias notables para estos tres lotes en el área bajo la curva de parasitemia versus tiempo, siendo de 43,9%; 58,6% y 64,6%, respectivamente. Estos últimos valores fueron muy importantes al momento de evaluar la sobrevida de los animales. Los animales control murieron todos entre los días 14 y 28 post-infección, mientras que el 83,3% de los animales tratados con Bnz suplementado con las vitaminas se mantuvieron con vida hasta el final del experimento (100 días post-infección). La vitamina B12 y el ácido ascórbico mostraron un efecto sinergístico sobre el efecto del Bnz solo.
En base a estos resultados, se propone como alternativa terapéutica para la enfermedad de Chagas, un tratamiento combinado B12+ácido ascórbico+Bnz, el cual permitiría disminuir la dosis de Bnz, reduciendo así los efectos colaterales perjudiciales que el mismo presenta.
Por otro lado, siguiendo con la aplicación de análogos estructurales al hemo como agentes antiparasíticos, surge la posibilidad de aplicar la inactivación fotodinámica (IFD) empleando porfirinas sintéticas como fotosensibilizantes. IFD es el nombre que recibe la TFD cuando se aplica a microorganismos, en lugar de cultivo de células o animales. En la actualidad este grupo está investigando la posibilidad de emplear IFD como técnica para el tratamiento antiparasitario de sangre contaminada con T. cruzi.
AGRADECIMIENTOS
Este grupo de trabajo destaca la eficiente colaboración de las Sras Victoria Castillo y Alejandra Caballero, de los Lics. Leda Oliveri, Pablo Winitzky y de los Dres. María del Carmen Martínez y Guillermo Noriega, todos ellos miembros de la Carrera del Personal de Apoyo a la Investigación del CONICET y/o docentes de la FCEN, UBA.
Así como expresar nuestro agradecimiento a todos los Investigadores, Personal de Apoyo, Becarios, Tesistas y Médicos que a lo largo de estos 38 años fueron o aún son miembros del CIPYP y han contribuido desde sus distintas posiciones, a la obtención de los resultados presentados en este trabajo. En especial a la memoria de la Dra. Susana Afonso, del Dr. Luis Espínola, del Sr. César Polo y de la Sra. Hilda Gasparoli.
Además, agradecer al CONICET; la Agencia de Promoción Científica y Tecnológica; la Universidad de Buenos Aires; la Municipalidad de la Ciudad de Buenos Aires; el Ministerio Nacional de Educación; el Banco de la Nación Argentina; la Secretaría de Estado de Salud Pública del Ministerio de Bienestar Social, el Ministerio de Salud Pública de la Nación; Laboratorios PROMECO SA, Buenos Aires; la International Association for Cancer Research, (IACR), UK; The Welcome Trust Foundation, UK; The University College London Medical School, UK; The British Council and The British Royal Society, UK; la Asociación Española para la Investigación Científica Internacional, España; el Instituto de Cultura Iberoamericano, ICI, España; el Ministerio de Educación y Ciencia, España; la DAAD, Alemania; The Paul S. Unna Foundation, Alemania; The University of Oslo, Noruega; The University of Marburg, Alemania; The Gordon Research Conferences, USA; por los subsidios otorgados para la financiación de estos trabajos; el intercambio de investigadores, becarios y doctorandos entre el CIPYP y varios Centros y Laboratorios de otros países en USA, Centro América, América del Sur, Europa, África, Asia y Australia; becas en y para el país y en y para el exterior, recepción de numerosos investigadores visitantes extranjeros así como la organización de tres Congresos Internacionales, como el Primer Congreso Internacional sobre Porfirinas y Porfirias (CIPYP), en el cual participaron especialistas de todos los Continentes y Terapia Fotodinámica del cáncer, en nuestro país. En estos casos, se publicaron todos los trabajos presentados en forma completa, en números especiales de las Revistas "International Journal of Biochemistry and Cell Biology", Batlle A. Editor, Elsevier, y el "Journal of Environmental Pathology, Toxicology and Oncology", Batlle A, Peng Q, Editors, Begel House, Inc. USA.
1. Batlle A. Porfirias Humanas. Signos y Tratamientos. En Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. Acta Bioquím Clín Latinoam 1997; Supl. 3; 37-69. [ Links ]
2. Maines MD, Gibbs PE. 30 some years of heme oxygenase: from a "molecular wrecking ball" to a "mesmerizing" trigger of cellular events. Biochem Biophys Res Commun 2005; 338: 568-77. [ Links ]
3. Sato H, Higashimoto, Sakamoto H, Sugishima M, Takahashi K, Palmer G, et al. Electrochemical reduction of ferrous α-verdoheme in complex with heme oxigenase-1. J Inorg Biochem 2007; 101: 1394-9.
4. Tomaro ML, Batlle AM. Bilirubin: its role in cytoprotection against oxidative stress. Review. Int J Biochem Cell Biol 2002; 34: 216-20. [ Links ]
5. Parera VE, Fukuda H, Stella AM, Wider E, Batlle AM del C. El saturnismo, una porfiria tóxica aguda, es una enfermedad toxogenética. Acta Bioquim Clin Latinoam 1986; 20 (3): 345-53. [ Links ]
6. Polo CF, Navone NM, Afonso SG, Buzaleh AM, Shoua E, Batlle A. Planificación de los estudios bioquímicos para un Laboratorio de Porfirinas. Acta Bioquím Clin Latinoam 1988; 22: 543-59. [ Links ]
7. Batlle A, Stella AM, Melito VA, Parera VE. Diez casos de Porfiria Cutánea Tardía infantil en la Argentina. Acta Bioquím Clin Latinoam 1989; 315: 20. [ Links ]
8. De Siervi A, Varela LS, Méndez M, Parera VE, Batlle AM del C, Rossetti MV. Estudios genéticos de las porfirias agudas en Argentina. Acta Bioquím Clín Latinoam 2002; 36 (5): 505-13. [ Links ]
9. Martinez M del C, Buzaleh AM, Batlle A. Efecto de la administración de los anestésicos Enflurano e Isoflurano a ratones con niveles inducidos y deprimidos de Citocromo P450. Estudios sobre el sistema enzimático metabolizante de drogas. Acta Bioquím Clin Latinoam 2005; 39: 37-42. [ Links ]
10. Melito VA, Parera VE, Rossetti MV, Batlle A. Manifestación de porfiria cutánea tardía en pacientes infectados con el virus de la Inmunodeficiencia humana. Acta Bioquím Clín Latinoam 2006; 40 (1): 29-34. [ Links ]
11. Rossetti MV, Parera VE, Melito VA, Batlle A. Porfiria Congénita Eritropoyética en la Argentina: Cuatro niños y un caso de manifestación tardía. Acta Bioquím Clín Latinoam 2007; 41 (3): 359-67. [ Links ]
12. Batlle A, Magnin P. Porfirias y Porfirinas. Aspectos Clínicos y Bioquímicos. Actualizaciones Médico Bioquímicas. Acta Bioquím Clín Latinoam Supl Nro. 2, 1988. [ Links ]
13. Melito VA, Rossetti MV, Parera VE, Batlle A. Porfirias poco frecuentes. Casos detectados en la población argentina. Rev Argent Dermatol 2006; 87: 248-63. [ Links ]
14. Cerbino GN, Gerez EN, Varela LS, Melito VA, Batlle A, Parera VE, et al Acute Intermittent Porphyria in Argentine. An Update. BioMed Res Int 2015; 946387. doi:10.1155/2015/946387. [ Links ]
15. De Siervi A, Mendez M, Parera VE, Varela LS, Batlle A, Rossetti MV. Acute intermittent porphyria: characterization of two novel mutations in the porphobilinogen deaminase gene, one amino acid deletion (453-455delAGC) and one splicing aceptor site mutation (IVS8-1G®T). Hum Mutat Mutation in Brief 1999; 14 (4): 355. [ Links ]
16. De Siervi A, Rossetti MV, Parera VE, Astrin KH, Aizencang GI, Glass IA, et al. Identification and characterization of Hydroxymethylbilane Synthase mutations causing Acute Intermittent Porphyria: Evidence for an ancestral founder of the common G111R mutation. Am J Med Genet 1999; 86: 366-75. [ Links ]
17. De Siervi A, Rossetti MV, Parera VE, Mendez M, Varela LS, Batlle AM del C. Acute intermittent porphyria: Biochemical and clinical analysis in the Argentinean population. Clin Chim Acta 1999; 288: 63-71. [ Links ]
18. De Siervi A, Weiss Cádiz DE, Parera VE, Batlle A, Rossetti MV. Identification and Characterization of two novel mutations that produce Acute Intermittent Porphyria: a 3-base deletion (841-843delGGA) and a missense mutation (T35M). Hum Mutat Mutation in Brief 2000; 16 (4): 373. [ Links ]
19. Parera VE, De Siervi A, Varela LS, Rossetti MV, Batlle A.M del C. Acute Porphyrias in the Argentinean population: A Review. Cell Mol Biol (Noisy-le-grand) 2003; 49 (4): 493-500. [ Links ]
20. Flagel MS, Parera VE, Batlle A, Rossetti MV. A novel duplication mutation in Argentinean Acute Intermittent Porphyria patients. Gene Bank 2011; HQ731552 [ Links ]
21. Flagel MS, Parera VE, Batlle A, Rossetti MV. Caracterización de una deleción intrónica en el gen de la PBGD para el diagnóstico presiintomático en Porfiria Aguda Intermitente [resumen]. Medicina 2012; 72 (Supl II): 234. [ Links ]
22. De Siervi A, Parera VE, Batlle A, Rossetti MV. Two new mutations (H106P and L178V) in the protoporphyrinogen oxidase gene in Argentinean patients with Variegate Porphyria. Hum Mutat Mutation and Polymorphism Report 2000; 16 (6): 532-34. [ Links ]
23. De Siervi A, Parera VE, Varela LS, Batlle A, Rossetti MV. A novel mutation (1320InsT) identified in two Argentine families with Variegate Porphyria. Hum Mutat Mutation and Polymorphism Report 2000; 16 (1): 96. [ Links ]
24. De Siervi A, Parera VE, Varela LS, Batlle A, Rossetti MV. Porfiria Variegata: signos cutáneos, agudos y bioquímicos. Primer estudio genético en pacientes argentinos. Rev Argent Dermatol 2000; 81: 94-100. [ Links ]
25. Rossetti MV, Granata BX, Giudice J, Parera VE, Batlle A. Genetic and biochemical studies in Argentinean patients with Variegate Porphyria. BioMed Central. Medical Genetics 2008; 9:54. doi:10.1186/1471-2350-9- 54. [ Links ]
26. Méndez M, Granata BX, Morán Jiménez MJ, Parera VE, Batlle A, Enríquez de Salamanca R, et al. Functional characterization of five protoporphyrinogen oxidase missense. Mutations found in Argentinean Variegate Porphyria patients. JIMD Reports 2011; doi:10.1007/8904_2011_77. [ Links ]
27. Granata BX, Baralle M, De Conti L, Parera VE, Rossetti, MV. Characterization of Variegate Porphyria mutations using a minigene approach. JIMD Reports 2014; doi:10.1007/8904_2014_388. [ Links ]
28. Granata BX, Parera VE, Batlle A, Rossetti MV. Haplotype study in Argentinean variegate porphyria patients. Hum Heredity 2016; 80(3):139-43. [ Links ]
29. Parera VE, Koole R, Batlle Alcira, & De Rooij Felix. Coproporphyria in Argentinean Patients: Genetic Studies [resumen]. 2003; Physiol Res 52: 19. [ Links ]
30. Medina NM, Colombo FP, Melito VA, Batlle A, Rossetti MV, Parera VE. Coproporfiria hereditaria: dos nuevas mutaciones en el gen de la coproporfirinógeno oxidasa [resumen]. Medicina 2013; 73 (Supll III): 280. [ Links ]
31. Siegesmund M, van Tuyll van Serooskerken AM, Poblete-Gutiérrez P, Frank J. The acute hepatic porphyrias: current status and future challenges. Best Pract Res Clin Gastroenterol 2010; 24(5): 593-605. [ Links ]
32. Anderson KE, Kappas A. The porphyrias. En: BC Decker inc (ed): Metabolism, 33(4). DOI: 10.2310/7900.1162, 2010. [ Links ]
33. Granata BX, Parera VE, Melito VA, Teijo MJ, Batlle AM, Rossetti MV. The very first description of a patient with hepatoerythropoietic porphyria in Argentina. Biochemical and molecular studies. Cell Mol Biol (Noisy-le-grand) 2009; 55 (1): 61-5. [ Links ]
34. Méndez M, Sorkin L, Rossetti MV, Astrin KH, Batlle A, Parera VE, et al. Familial Porphyria Cutanea Tarda: Characterization of seven novel Uroporphyrinogen Decarboxylase gene mutations and frequency of common Hemochromatosis alleles. Am J Hum Genet 1998; 63: 1365-75. [ Links ]
35. Méndez M, Rossetti MV, De Siervi A, Batlle A, Parera VE. Mutations in Familial Porphyria Cutanea Tarda: two novel and two previously described for Hepatoerythropoietic Porphyria. Hum Mut Mutation in Brief 2000; 16 (3): 269-70. [ Links ]
36. Méndez M, Rossetti MV, Batlle A, Parera VE. The role of inherited and acquired factors in the development of Porphyria Cutanea Tarda in the Argentinean population. J Am Acad Dermatol 2005; 52 (3): 417-24. [ Links ]
37. Méndez M, Rossetti MV, Gómez-Abecia S, Morán- Jiménez MJ, Parera VE, Batlle A, et al, Molecular analysis of the UROD gene in 17 Argentinean patients with familial porphyria cutanea tarda: Characterization of four novel mutations. Mol Genet Metabol 2012; 105 (4): 629-33. [ Links ]
38. Medina NM, Colombo FP, Piñeiro Pauwels MB, Rossetti MV, Batlle A, Parera VE. Detección de 4 nuevas mutaciones en el gen UROD en familias con Porfiria Cutánea Tardía [resumen]. Medicina 2012; 72 (Supl II): 234. [ Links ]
39. Medina NM, Colombo FP, Rossetti MV, Batlle A, Parera VE. Four novel UROD mutations identified in Argentinean patients with familial Porphyria Cutanea Tarda [resumen]. Clin Chem Lab Med 2013; 51(5): eA1-eA64. [ Links ]
40. Medina NM, Granata BX, Cerbino GN, Colombo FP, Rossetti MV, Batlle A, et al. Gen UROD: Estudio molecular y diagnóstico diferencial de Porfiria [resumen]. J Basic Appl Gen 2013; 24 (1): 136. [ Links ]
41. Colombo FP, Rossetti MV, Mendez M, Martínez JE, Enríquez de Salamanca R, Batlle A et al, Functional associations of genetic variants involved in the clinical manifestation of erythropoietic protoporphyria in the Argentinean Population. J Eur Dermatol Vennereol 2012; doi:10.1111/j.1468-3083.2012.04566.x. [ Links ]
42. Gouya L, Martin-Schmitt C, Robreau AM, Austerlitz F, Da Silva V, Brun P. Contribution of a common single nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am J Hum Genet 2006; 78: 2-14. [ Links ]
43. Gouya L, Puy H, Lamoril J, Da Silva V, Grandchamp B, Nordmann Y, et al. Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood 1999; 93 (6): 2105-10. [ Links ]
44. Gouya L, Puy H, Robreau AM, Bourgeois M, Lamoril J, Da Silva V, et al. The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wildtype FECH. Nat Genet 2002; 30 (1): 27-8. [ Links ]
45. Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet 2008; 83 (3): 408-14. [ Links ]
46. Parera VE, Koole RH, Minderman G, Rossetti MV, Batlle A, De Rooij F. Novel null–allele mutations and genotype-phenotype correlation in Argentinean patients with Erythropoietic Protoporphyria. Mol Med 2009; 15: 425-31.
47. Deybach JC, de Verneuil H, Phung N, Nordmann Y, Puissant A, Boffety BJ. Congenital erythropoietic porphyria (Günther’s disease): enzymatic studies in two cases of late onset. Lab Clin Med 1981; 97 (4): 551-8.
48. Piñeiro Pauwels MB, Gerez EN, Melito VA, Martínez M, Batlle A, Parera V et al. Transplante de médula ósea como tratamiento en la Porfiria Congénita Eritropoyética. Evolución del cuadro bioquímico y genético [resumen]. Medicina 2009; 69 (Supl I): 101. [ Links ]
49. Schüller A, Salamanca R. Metabolismo de las Porfirinas y Porfirias. Madrid: Ed. Complutense, 2000. [ Links ]
50. Albers JW, Fink JK. Porphyric neuropathy. Muscle Nerve 2004; 30(4): 410-22. [ Links ]
51. Pischik E, Kauppinen R. Neurological manifestations of acute intermittent porphyria, Cell Mol Biol (Noisy-le-grand) 2009; 55(1): 72-83. [ Links ]
52. Besur S, Hou W, Schmeltzer P, Bonkovsky H. Clinically Important features of porphyrin and heme metabolism and the porphyrias. Metabolites 2014; 4(4): 977- 1006. [ Links ]
53. Herrick AL, McColl KEL. Acute intermittent porphyria. Best Practice Res. Clin Gastroenterol 2006; 19: 135- 249. [ Links ]
54. Princ F, Juknat A, Batlle A. Porphyrinogenesis in rat cerebellum. Effect of high delta-aminolevulinic acid concentration. Gen Pharmacol 1994; 25: 761-6. [ Links ]
55. Stummer W, Stocker S, Novotny A, Heimann A, Sauer O, Kempski O, et al. In vitro and in vivo porphyrin accumulation by C6 glioma cells after exposure to 5-aminolevulinic acid. J Photochem Photobiol 1008; 45: 160-9. [ Links ]
56. Bechara E. Oxidative stress in acute intermittent porphyria and lead poisoning may be triggered by 5-aminolevulinic acid. Braz J Med Biol Res 1996; 29: 841-51. [ Links ]
57. Demasi M, Penalti C, DeLucia R, Bechara E. The prooxidant effect of 5-aminolevulinic acid in the brain tissue of rats: implications in neuropsychiatric manifestations in porphyrias. Free Rad Biol Med 1996; 20: 291-9. [ Links ]
58. Ennis S, Novotny A, Xiang J, Shakui P, Masada T, Stummer W, et al. Transport of 5-aminolevulinic acid between blood and brain. Brain Res 2003; 959: 226-34. [ Links ]
59. Adhikari A, Penatti C, Resende R, Ulrico H, Britto L, Bechara E. 5- aminolevulinate and 4,5-dioxovalerate ions decrease GABA(A) receptor density in neuronal cells, synaptosomes and rat brain. Brain Res 2006; 1093: 95-104. [ Links ]
60. Emanuelli T, Pagel F, Porciuncula L, Souza D. Effects of 5-aminolevulinic acid on the glutamatergic neurotransmission. Neurochem Int 2003; 42: 115-21. [ Links ]
61. Lavandera J, Rodríguez J, Ruspini S, Martínez M.del C, Gerez E, Batlle A, et al. Pleiotropic effects of 5-aminolevulinic acid in mice brain. Biochem.Cell Biol 2016; 94: 1-9. http://dx.doi.org/10.1139/bcb-2015-0094. [ Links ]
62. Quail A. Modern inhalation anaesthetic agents. A review of halothane, isoflurane and enflurane. Med J Austr 1989; 150: 95-102. [ Links ]
63. Behne M, Wilke HJ, Harder S. Clinical pharmacokinetics of sevoflurane. Clin Pharmacokinet 1999; 36: 13-26. [ Links ]
64. Kharasch E, Thummel K. Identification of cytochrome P-450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane and methoxyflurane. Anesthesiology 1993; 79: 795-807. [ Links ]
65. Buzaleh AM, Enríquez de Salamanca R, Batlle AM. Porphyrinogenic properties of the anesthetic enflurane. Gen Pharmac 1992; 23: 665-9. [ Links ]
66. Buzaleh AM, Enríquez de Salamanca R, Batlle AM. Administration of the anesthetic isoflurane to mice: a model for acute intermittent porphyria? J Pharmacol Toxicol Methods 1992; 28(4): 191-7. [ Links ]
67. Buzaleh AM, Enríquez de Salamanca R, Batlle A. Effect of multiple doses of volatile anesthetics on heme enzymes. Gen Pharmac 1994; 25: 1179-83. [ Links ]
68. Buzaleh AM, Vazquez E, Nuñez G, Batlle A. Effect of chronic anesthesia on the drug metabolizing enzyme system and heme pathway regulation. Gen Pharmac 1997; 28: 577-82. [ Links ]
69. Sampayo R, Lavandera J, Batlle A, Buzaleh AM. Sevoflurane: its action on heme metabolism and Phase I drug metabolizing system. Cell Mol Biol (Noisy-le-grand) 2009; 55 (2): 134-40. [ Links ]
70. Buzaleh AM, Batlle A. Strain and sex differences in the effect of enflurane and isoflurane on heme metabolism in mice. Gen Pharmac 1996; 27: 1009-12. [ Links ]
71. Buzaleh AM, Vazquez E, Batlle A. Alterations in fluorinated ether anesthetics effects on heme metabolism following chronic ethanol consumption. Gen Pharmac 1995; 26: 1425-29. [ Links ]
72. Buzaleh AM, Martínez M del C, Batlle A. Relevance of cytochrome P450 levels in the actions of enflurane and isoflurane in mice. Studies on the haem pathway. Clin Exp Pharmacol Physiol 2000; 27: 796-800. [ Links ]
73. Buzaleh AM, Batlle A. Glutathione depletion and anaesthesia in mice alter heme and drug metabolising enzymes. Biochim Biophys Acta 2005; 1723: 128-34. [ Links ]
74. Buzaleh AM, Batlle A. Administration of volatile anaesthetics to animals treated with cobalt as inductor of heme oxygenase. Pharmacology Rev Communications 2000; 11: 39-47. [ Links ]
75. Lindberg RL, Porcher C, Grandchamp B, Ledermann B, Bürki K, Brandner S, et al. Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat Genet 1996; 12(2): 195-9. [ Links ]
76. Ruspini S, Zuccoli J, Lavandera J, Batlle A, Buzaleh AM. Efecto de los anestésicos Isoflurano y Sevoflurano sobre la biosíntesis del hemo en un modelo murino de Porfiria Aguda Intermitente [resumen]. Medicina 2012; 72 (Supl. II): 121. [ Links ]
77. Buzaleh AM, García Bravo M, Navarro S, Moran Jimenez MJ, Méndez M, Batlle A, et al. Volatile anaesthetics induce biochemical alterations in the heme pathway in a B-lympocyte cell line established from hepatoerythropoietic porphyria patients (LBHEP) and in mice inoculated with LBHEP cells. Int J Biochem Cell Biol 2004; 36: 216-22. [ Links ]
78. Buzaleh AM, Morán-Jiménez MJ, García-Bravo M, Sampedro A, Batlle A, Enríquez de Salamanca R, et al. Induction of hepatic aminolevulinate acid synthetase activity by isoflurane in a genetic model for erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand) 2009; 55 (1): 38-44. [ Links ]
79. Buzaleh AM, Moreno-Carralero MI, Méndez M, Batlle A, Enríquez de Salamanca R, Morán-Jimenez MJ. Heme metabolism, oxidative and nitrosative markers in a mouse model of hemochromatosis. Effect of isoflurane, ethanol and 5-aminolevulinic acid. J Pathol Res 2013; 2: 15-20. [ Links ]
80. Rodríguez JA, Buzaleh AM, Fossati M, Azcurra J, Batlle AM del C. The effects of some porphyrinogenic drugs on the brain cholinergic system. Cell Mol Biol (Noisy-le-grand) 2002; 48 (1): 103-10. [ Links ]
81. Lavandera J, Fossatti M, Azcurra J, Batlle A, Buzaleh AM. Glutamatergic System: another target for the action of porphyrinogenic agents. Cell Mol Biol (Noisy-le-grand) 2009; 55 (1): 23-8. [ Links ]
82. Rodriguez JA, Martinez M del C, Gerez E, Batlle A, Buzaleh AM. Heme oxygenase, aminolevulinate acid synthetase and the antioxidant system in the brain of mice treated with porphyrinogenic drugs. Cell Mol Biol (Noisy-le-grand) 2005, 51: 487-94. [ Links ]
83. Lavandera J, Batlle A, Buzaleh AM. Metabolization of porphyrinogenic agents in brain: involvement of the Phase I drug metabolizing system. A comparative study in liver and kidney. Cell Mol Neurobiol 2007; 27: 717-29. [ Links ]
84. Lavandera J, Ruspini S, Batlle A, Buzaleh AM. Cytochrome P450 expression in mouse brain: specific isoenzymes involved in phase I metabolizing system of porphyrinogenic agents in both microsomes and mitochondria. Biochem Cell Biol 2014; 92: 1-6. http://dx.doi.org/10.1139/bcb-2014-0088. [ Links ]
85. Lavandera J, Batlle A, Buzaleh AM. Mice brain nitric oxide synthase response induced by anaesthetics and other porphyrinogenic drugs. Drug Metab Lett 2011; 5: 25-9. [ Links ]
86. Buzaleh AM, Meiss R, Lavandera J, Vallecorsa P, Ruspini S, Batlle A. Óxido nítrico sintasa y hemo oxigenasa en encéfalo de ratones tratados con anestésicos volátiles y otros agentes porfirinogénicos. Estudio inmunohistoquímico de la expresión proteica [resumen]. Medicina 2012; 72 (Supl. II): 121. [ Links ]
87. Zuccoli J, Ruspini S, Batlle A, Buzaleh AM. Alteración en las actividades enzimáticas de los complejos de la cadena respiratoria mitocondrial (I-III, II-III y IV) por efecto de agentes porfirinogénicos [resumen]. Medicina 2013; 73 (Supl I): 256. [ Links ]
88. Batlle A, Llambías EBC, Wider de Xifra EA, Tigier HA. Porphyrin biosynthesis in soybean callus system XV. The effect of growth conditions. Int J Biochem 1975; 6: 591-606. [ Links ]
89. Fukuda H, Casas A, Batlle A. Aminolevulinic acid: from its unique biological function to its star role in photodynamic therapy. Int J Biochem Cell Biol 2005; 37: 272-6. [ Links ]
90. Fukuda H, Casas A, Batlle A. Use of ALA and ALA derivatives for optimizing ALA-PDT. A Review of our experience. J Envir Pathol Toxicol Oncol 2006; 25: 127- 43. [ Links ]
91. Fukuda H, Paredes SR, Batlle AM del C. Tumor localizing properties of porphyrins. In vivo studies using free and liposome encapsulated aminolevulinic acid. Comp Biochem Physiol 1992; 102B: 433-6. [ Links ]
92. Alvarez MG, Lacelli MS, Rivarola V, Batlle A, Fukuda H. 5-aminolevulinic acid-mediated photodynamic therapy on Hep-2 and MCF-7c3 cells. J Env Pathol Toxicol Oncol 2007; 26 (2): 75-82. [ Links ]
93. Alvarez MG, Milanesio M, Rivarola V, Durantini E, Batlle A, Fukuda H. Endogenous and exogenous porphyrins as photosensitizers in the HEP-2 human carcinoma line. Cell Mol Biol (Noisy-le-grand) 2009; 55 (2): 8-14. [ Links ]
94. Hrkal Z, Grebenová D, Cajthamlová H, Soucek J, Bartosová J, Fuchs O, et al. Use of photodynamic therapy for elimination of residual leukemic cells in autologous transplants of hematopoietic progenitor cells. Cas Lek Cesk 2002; 141 (Suppl): 41-6. [ Links ]
95. Huang HF, Chen YZ, Wu Y, Chen P. Purging of murine erythroblastic leukemia by ZnPcS2P2-based-photodynamic therapy. Bone Marrow Transplant 2006; 37 (2): 213-7. [ Links ]
96. Capella MA, Capella LS. A light in multidrug resistance: photodynamic treatment of multidrug-resistant tumors. J Biomed Sci 2003; 10 (4): 361-6. [ Links ]
97. Chu ES, Yow CM, Shi M, Ho RJ. Effects of photoactivated 5-aminolevulinic acid hexyl ester on MDR1 over-expressing human uterine sarcoma cells. Toxicol Lett 2008; 181 (1): 7-12. [ Links ]
98. Tang PM, Zhang DM, Xuan NH, Tsui SK, Waye MM, Kong SK et al. Photodynamic therapy inhibits P-glycoprotein mediated multidrug resistance via JNK activation in human hepatocellular carcinoma using the photosensitizer pheophorbide a. Mol Cancer 2009; 8: 56. [ Links ]
99. Diez B, Cordo Russo R, Teijo MJ, Hajos S, Batlle A, Fukuda H. ROS production by endogenously generated Protoporphyrin IX in murine leukemia cells. Cell Mol Biol (Noisy-le-grand) 2009; 55 (2): 15-9. [ Links ]
100. Diez B, Ernst G, Teijo MJ, Batlle A, Hajos S, Fukuda H. Combined Chemotherapy and ALA based Photodynamic Therapy in leukemic murine cells. Leukemia Res 2012; 36: 1179-84. [ Links ]
101. Canti B, Nicolin A, Cubeddu R, Taroni P, Bandieramonte G, Valentini G. Antitumor efficacy of the combination of photodynamic therapy and chemotherapy in murine tumors. Cancer Lett 1998; 125: 39-44. [ Links ]
102. Snyder JW, Greco WR, Bellnier DA, Vaughan L, Henderson BW. Photodynamic therapy: a means to enhanced drug delivery to tumors. Cancer Res 2003; 63 (23): 8126-31. [ Links ]
103. Kirveliene V, Grazeliene G, Dabkeviciene D, Micke I, Kirvelis D, Juodka B et al. Schedule-dependent interaction between Doxorubicin and mTHPC-mediated photodynamic therapy in murine hepatoma in vitro and in vivo. Cancer Chemother Pharmacol 2006; 57 (1): 65-72. [ Links ]
104. Anand S, Honari G, Hassan T, Elson P, Maytin EV. Low-dose methotrexate enhances aminolevulinate-based photodynamic therapy in skin carcinoma cells in vitro and in vivo. Clin Cancer Res 2009; 15 (10): 3333-43. [ Links ]
105. Xie Q, Jia L, Liu YH, Wei CG. Synergetic anticancer effect of combined gemcitabine and photodynamic therapy on pancreatic cancer in vivo. World J Gastroenterol 2009; 15 (6): 737-41. [ Links ]
106. Diez B, Hajos S, Meiss R, Ernst G, Teijo MJ, Batlle A et al. In vitro and In vivo 5-Aminolevulinic Acid-Based Photodynamic Therapy in Sensitive and Drug Resistant Leukemic Murine Cells. En: Photodynamic Therapy: Fundamentals, Applications and Health Outcome Effects. Ed Adrian Hugo, Hauppauge, NY, Cap 7: p.173- 91; 2015. [ Links ]
107. Canti G, Calastretti A, Bevilacqua A, Reddi E, Palumbo G, Nicolin A. Combination of photodynamic therapy + immunotherapy + chemotherapy in murine leukemia. Neoplasma 2010; 57 (2): 184-8. [ Links ]
108. Cassinelli J, Teijo MJ, Rossetti MV, Fukuda H, Batlle A. Porphyrin biosynthesis and photosensitization in human and murine lung adenocarcinoma cell lines. Clin Chem Lab Med 2013; 51 (5): eA51, P51. [ Links ]
109. Teijo MJ, Meiss R, Diez B, Batlle A, Fukuda H. Reactive Oxygen Species as main factors conditioning cell death in response to ALA-based Photodynamic Therapy in human lung adenocarcinoma spheroids J Thor Oncol 2012; 7 (7) (Suppl 2): S101-S156. [ Links ]
110. Fukuda H, Teijo MJ, Batlle A. Human lung adenocarcinoma cells sensitive and resistant to ALA-based Photodynamic Therapy [resumen]. J Thor Oncol 2014; 9 (9) (Suppl 3): S175. [ Links ]
111. Teijo MJ, Diez B, Batlle A, Fukuda H. Modulation of 5-Aminolevulinic acid mediated photodynamic therapy induced cell death in a human lung adenocarcinoma cell line, ICST 2016; doi:10.15761/ICST.1000187. [ Links ]
112. Salzman TA, Stella AM, Wider de Xifra EA, Batlle AM del C, Docampo R, Stoppani AOM. Porphyrin biosynthesis in parasitic hemoflagellates: Functional and defective enzymes in Trypanosoma cruzi. Comp Biochem Physiol 1982; 72B: 663-7. [ Links ]
113. Salzman TA, Batlle AM del C, De Souza W. Heme synthesis in Trypanosoma cruzi: Influence of the strain and culture medium. Comp Biochem Physiol 1986; 83B: 57-61. [ Links ]
114. Lombardo ME, Araujo LS, Batlle A. 5-Aminolevulinic acid synthesis in epimastigotes of Trypanosoma cruzi. Int J Biochem Cell Biol 2003; 35: 1263-71. [ Links ]
115. Beale SI, Castelfranco PA. The biosynthesis of δ-aminolevulinic acid in higher plants. II. Formation of 14C-δ-aminolevulinic acid from labeled precursors in greening plant tissues. Plant Physiol 1974; 53: 297-303.
116. Salvador GF. La Synthese d’acide δ-aminolevulinique par des chloroplastes isoles d’Euglena gracilis. C R Acad Sci Paris; 286: 49-52; 1978.
117. Lombardo ME, Araujo LS, Juknat AA, Batlle AM del C. Effect of illumination on growth, chlorophyll content and delta-aminolevulinic acid synthesis in Euglena gracilis. Comp Biochem Physiol 1988; 91B: 279-84. [ Links ]
118. Lombardo ME, Araujo LS, Juknat AA, Batlle AM del C. Glutamate: 4,5-dioxovaleric acid transaminase from Euglena gracilis. Kinetic studies. Eur J Biochem 1989; 182: 657-60. [ Links ]
119. Lombardo ME, Araujo LS, Batlle AM del C. Kinetic studies on the glutamate: glyoxylate transaminase of Euglena gracilis. J Photochem Photobiol 1996; 36: 241-4. [ Links ]
120. Salzman TA, Batlle AM del C. Heme synthetase in Trypanosoma cruzi. Comp Biochem Physiol 1986; 85B: 537-41. [ Links ]
121. Ciccarelli A, Araujo L, Batlle A, Lombardo E. Effect of haemin on growth, protein content and the antioxidant defence system in Trypanosoma cruzi. Parasitology 2007; 134: 959-65. [ Links ]
122. Chen J, London I. Regulation of protein synthesis by heme-regulated eIF-2α kinase. Trends Biochem Sci 1995; 20: 105-8.
123. Pal J, Joshi-Purandare M. Dose-dependent differential effect of hemin on protein synthesis and cell proliferation in Leishmania donovani promastigotes cultured in vitro. J Biosci 2001; 26: 225-31. [ Links ]
124. Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicol Lett 2005; 157: 175-88. [ Links ]
125. Turrens JF. Oxidative stress and antioxidant defenses: a target for the treatment of diseases caused by parasitic protozoa. Mol Aspects Med 2004; 25: 211-20. [ Links ]
126. Motterlini R, Green C, Foresti R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid Redox Signal 2002; 4: 615-24. [ Links ]
127. Ciccarelli A, Frank F, Puente V, Malchiodi E, Batlle A, Lombardo ME. Anti-parasitic effect of vitamin B12 on Trypanosoma cruzi. Antimicrob Agents Chemother 2012; 56: 5315-20. [ Links ]
128. Solovieva ME, Soloviev VV, Akatov VS. Vitamin B12b increases the cytotoxicity of short-time exposure to ascorbic acid, inducing oxidative burst and iron-dependent DNA damage. Eur J Pharmacol 2007; 566: 206-14. [ Links ]
129. Solovieva ME, Soloviev VV, Kudryavtsev AA, Trizna YA, Akatov VS. Vitamin B12b enhances the cytotoxicity of dithiothreitol. Free Radic Biol Med 2008; 44: 1846- 56. [ Links ]
130. Kauppinen R, Mustajoki S, Pihlaja H, Peltonen L, Mustajoki P. Acute intermittent porphyria in Finland: 19 mutations in the porphobilinogen deaminase gene. Hum Mol Genet 1995; 4 (2): 215-22. [ Links ]
131. Lundin G, Hashemi J, Floderus Y, Thunell S, Sagen E, Laegreid A et al. Four mutations in the porphobilinogen deaminase gene in patients with acute intermittent porphyria. J Med Genet 1995; 32 (12): 979-81. [ Links ]
132. Gregor A, Schneider-Yin X, Szlendak U, Wettstein A, Lipniacka A, Rüfenacht UB et al. Molecular study of the hydroxymethylbilane synthase gene (HMBS) among Polish patients with acute intermittent porphyria. Hum Mut 2002; 19 (3): 310. [ Links ]
133. Luchinina YA, Surin VL, Luk’yanenko AV, Karpova IV, Pustovoit YS, Kravchenko SK. Molecular diagnostics of acute intermittent porphyria in Russia [Resumen] Eur J Hum Genet 1995; 13 (Suppl 1): 134.
134. Hessels J, Voortman G, van der Wagen A, van der Elzen C, Scheffer H, Zuijderhoudt FM. Homozygous acute intermittent porphyria in a 7-years-old boy with massive excretions of porphyrins and porphyrin precursors. J Inherit Metab Dis 2004; 27: 19-27. [ Links ]
135. Gu XF, de Rooij F, de Baar E, Bruyland M, Lissens W, Nordmann Y et al. Two novel mutations of the porphobilinogen deaminase gene in acute intermittent porphyria. Hum Mol Genet 1993; 2 (10): 1735-6. [ Links ]
136. Cappellini MD, Martinez di Montemuros F, Di Pierro E, Fiorelli G. Hematologically important mutations: acute intermittent porphyria. Blood Cells Mol Dis 2002; 28 (1): 5-12. [ Links ]
137. Gerez E, Cerbino G, Colombo F, Parera V, Batlle A, Rossetti MV. A novel splicing mutation (C.450-2A>G) in an Argentinean family with Acute Intermittent Porphyria. GENE BANK 2011; [ Links ] HM856802.
138. Delfau MH, Picat C, De Rooij F, Voortman G, Deybach JC, Nordmann Y et al. Molecular heterogeneity of acute intermittent porphyria: identification of four additional mutations resulting in the CRIM-negative subtype of the disease. Am J Hum Genet 1991; 49 (2): 421-8. [ Links ]
139. Delfau MH, Picat C, De Rooij FW, Hamer K, Bogard M, Wilson JH, et al. Two different point G to A mutations in exon 10 of the porphobilinogen deaminase gene are responsible for acute intermittent porphyria. J Clin Invest 1990; 86 (5): 1511-6. [ Links ]
140. Kauppinen R, Peltonen L, Pihlaja H, Mustajoki P. CRIM-positive mutations of acute intermittent porphyria in Finland. Hum Mutat 1992; 1 (5): 392-6. [ Links ]
141. Lundin G1, Wedell A, Thunell S, Anvret M. Two new mutations in the porphobilinogen deaminase gene and a screening method using PCR amplification of specific alleles. Hum Genet 1994; 93 (1): 59-62. [ Links ]
142. Mgone CS, Lanyon WG, Moore MR, Louie GV, Connor JM. Identification of five novel mutations in the porphobilinogen deaminase gene. Hum Mol Genet 1994; 3 (5): 809-11. [ Links ]
143. Mgone CS, Lanyon WG, Moore MR, Louie GV, Connor JM. Detection of a high mutation frequency in exon 12 of the porphobilinogen deaminase gene in patients with acute intermittent porphyria. Hum Genet 1993; 92 (6): 619-22. [ Links ]
144. Puy H, Deybach JC, Lamoril J, Robreau AM, Nordmann. Detection of four novel mutations in the porphobilinogen deaminase gene in French Caucasian patients. with acute intermittent porphyria. Hum Hered 1996; 46 (3): 177-80. [ Links ]
145. Puy H, Deybach JC, Lamoril J, Robreau AM, Silva VD, Gouya L et al. Molecular epidemiology and diagnosis of PBGD gene defects in AIP. Am J Hum Genet 1997; 60: 1373-83. [ Links ]
146. Floderus Y, Shoolingin-Jordan PM, Harper P. Acute intermittent porphyria in Sweden. Molecular, functional and clinical consequences of some new mutations found in the porphobilinogen deaminase gene. Clin Genet 2002; 62 (4): 288-97. [ Links ]
147. Schuurmans MM, Schneider-Yin X, Rüfenacht UB, Schnyder C, Minder CE, Puy H et al Influence of age and gender on the clinical expression of acute intermittent porphyria based on molecular study of porphobilinogen deaminase gene among Swiss patients. Mol Med 2001; 7 (8): 535-42. [ Links ]
148. Wiman A, Harper P, Floderus Y. Nine novel mutations in the protoporphyrinogen oxidase gene in Swedish families with variegate porphyria. Clin Genet 2003; 64 (2): 122-30. [ Links ]
149. Granata BX. Porfirias Hepáticas Agudas. Relación con el estrés oxidativo y patologías asociadas (Tesis de Doctorado), UBA; 2013. [ Links ]
150. Whatley SD, Puy H, Morgan RR, Robreau AM, Roberts AG, Nordmann Y et al. Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet 1999; 65 (4): 984-94. [ Links ]
151. Deybach JC, Puy H, Robréau AM, Lamoril J, Da Silva V, Grandchamp B et al. Mutations in the protoporphyrinogen oxidase gene in patients with variegate porphyria. Hum Mol Genet 1996; 5 (3): 407-10. [ Links ]
152. Frank J, Jugert FK, Merk HF, Kalka K, Goerz G, Anderson K et al. A spectrum of novel mutations in the protoporphyrinogen oxidase gene in 13 families with variegate porphyria. J Invest Dermatol 2001; 116 (5): 821-3. [ Links ]
153. Cerbino G, Rossetti MV, Batlle A, Parera VE. Porfiria Cutánea Tardía. Diagnóstico molecular en pacientes argentinos [resumen]. J Basic Appl Genetics 2014; XXV (Supl I): 111. [ Links ]
154. McManus JF, Begley CG, Sassa S, Ratnaike S. Five new mutations in the uroporphyrinogen decarboxylase gene identified in families with cutaneous porphyria. Blood 1996; 88 (9): 3589-600. [ Links ]
155. Morán Jiménez MJ, Ged C, Romana M, Enríquez De Salamanca R, Taïeb A, Topi G et al. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet 1996; 58: 712- 21. [ Links ]
156. Badenas C, To-Figueras J, Phillips JD, Warby CA, Muñoz C, Herrero C. Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives. Clin Genet 2009; 75 (4): 346-53. doi: 10.1111/j.1399-0004.2009.01153.x [ Links ]
157. Meguro K, Fujita H, Ishida N, Akagi R, Kurihara T, Galbraith RA et al. Molecular defects of uroporphyrinogen decarboxylase in a patient with mild hepatoerythropoietic porphyria. J Invest Dermatol 1994; 102 (5): 681-5. [ Links ]
158. Brancaleoni V, Di Pierro E, Besana V, Ausenda S, Cappellini MD. Novel human pathological mutations. Gene symbol: UROD. Disease: porphyria, cutaneous. Hum Genet 2007; 122 (5): 554-5. [ Links ]
159. Brady JJ, Jackson HA, Roberts AG, Morgan RR, Whatley SD, Rowlands GL et al. Co-inheritance of mutations in the uroporphyrinogen decarboxylase and hemochromatosis genes accelerates the onset of porphyria cutanea tarda. J Invest Dermatol 2000; 115 (5): 868-74. [ Links ]
160. Wang X, Piomelli S, Peacocke M, Christiano AM, Poh-Fitzpatrick MB. Erythropoietic protoporphyria: four novel frameshift mutations in the ferrochelatase gene. J Invest Dermatol 1997; 109 (5): 688-91. [ Links ]
161. Henriksson M, Timonen K, Mustajoki P, Pihlaja H, Tenhunen R, Peltonen L, et al. Four novel mutations in the ferrochelatase gene among erythropoietic protoporphyria patients. J Invest Dermatol 1996; 106: 346–50.
162. Schneider-Yin X, Schäfer BW, Tönz O, Minder EI. Human ferrochelatase: a novel mutation in patients with erythropoietic protoporphyria and an isoform caused by alternative splicing. Hum Genet 1995; 95 (4): 391-6. [ Links ]
163. Frank J, Nelson J, Wang X, Yang L, Ahmad W, Lam H et al. Erythropoietic protoporphyria: identification of novel mutations in the ferrochelatase gene and comparison of biochemical markers versus molecular analysis as diagnostic strategies. J Investig Med 1999; 47: 278-84. [ Links ]
164. Imoto S, Tanizawa Y, Sato Y, Kaku K, Oka Y. A novel mutation in the ferrochelatase gene associated with erytropoietic protoporphyria. Br J Haematol 1996; 94: 191-7. [ Links ]
165. Di Pierro E, Moriondo V, Cappellini MD. Gene Symbol: FECH. Disease: Porphyria, erythropoietic. Hum Genet 2004; 114 (6): 609. [ Links ]
166. Melito V, Colombo F, Granata BX, Cano G, Infante L, Batlle A, Rossetti MV, Parera VE. Nueva variante mutacional del gen FECH asociada a un caso grave de Porfiria Cutánea atípica [resumen]. Medicina 2011; 71 (Supl III): 67. [ Links ]
167. Di Pierro E, Brancaleoni V, Moriondo V, Besana V, Cappellini MD. Co-existence of two functional mutations on the same allele of the human ferrochelatase gene in erythropoietic protoporphyria. Clin Genet 2007; 71: 84–8.
168. Colombo FP, Martinez JE, Rossetti MV, Batlle A, Parera VE. Prevalencia de mutaciones nonsense en pacientes con Protoporfiria Eritropoyética [resumen]. LILLOA 2009; 45 (Supl 1): 142. [ Links ]
Recibido: 12 de julio de 2016
Aceptado: 21 de septiembre de 2016

