










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.50 no.4 La Plata dic. 2016
BIOQUÍMICA CLÍNICA
Validación de la determinación de manganeso, molibdeno y zinc por plasma de acoplamiento inductivo
Validation of the determination of manganese, molibdenum and zinc using inductively coupled plasma
Validação da determinação de manganês, molibdênio e zinco por plasma de acoplamento indutivo
Silvia Sara Farías1a, Ana María Menéndez2b, Rodrigo Martín Pontiggia3b,c, Roberto Enrique Servant4a, Iris Cortez5c, María Luz Pita Martín de Portela6d
1 Dra. en Ciencias Químicas, Universidad de Buenos Aires.
2 Dra. de la Universidad de Buenos Aires, área Nutrición.
3 Dr. de la Universidad de Buenos Aires, área Química Biológica.
4 Lic. en Ciencias Químicas.
5 Técnica Química.
6 Dra. en Farmacia y Bioquímica, Universidad de Buenos Aires.
a Gerencia de Química. Centro Atómico Constituyentes. Comisión Nacional de Energía Atómica Av. General Paz 1499. San Martín, Provincia de Buenos Aires, Argentina.
b Facultad de Ciencias Exactas y Naturales y Ciencias de la Salud, Carrera de Farmacia. Universidad de Belgrano, Villanueva 1324. C1426DQG, Ciudad Autónoma de Buenos Aires, Argentina.
c Universidad de Belgrano, Villanueva 1324. C1426DQG, Ciudad Autónoma de Buenos Aires, Argentina & Benito Roggio Ambiental SA, Salguero 3800 (1407), Ciudad Autónoma de Buenos Aires, Argentina.
d Universidad de Buenos Aires. Facultad de Farmacia y Bioquímica: Departamento de Sanidad, Nutrición, Bromatología y Toxicología, Cátedra de Nutrición. Buenos Aires, Argentina.
CORRESPONDENCIA Dra. SILVIA SARA FARÍAS Sucre 4480 1430BWX CIUDAD AUTÓNOMA DE BUENOS AIRES E-mail: fariassilvias@gmail.com, farias@cnea.gov.ar
Resumen
Se describe la validación de un método para la determinación de manganeso, molibdeno y zinc, a niveles traza, en soluciones acuosas, mediante espectroscopia de emisión-plasma inductivo de argón. Se optimizó y validó la cuantificación de manganeso, molibdeno y zinc en solución acuosa ácida usando un espectrómetro de emisión atómica de plasma inductivo. Se determinaron: selectividad/especificidad, linealidad, repetibilidad y precisión intermedia utilizando materiales de referencia, y sesgo contrastando contra material de referencia certificado de matriz. Las longitudes de onda (nm) seleccionadas fueron: Zn 213.857, Mn 257.610 y Mo 202.031, las cuales permitieron discriminar interferencias espectrales. Se probó la linealidad de las funciones respuesta mediante evaluaciones estadísticas ad hoc. La precisión intermedia varió entre 5 y 11% y el sesgo no superó el 12%. Los límites de cuantificación (μg/L) fueron: Mn: 5; Zn: 10 y Mo: 10. Las incertidumbres asociadas a las determinaciones oscilaron entre 10 y 16%. La validación del método propuesto demostró que es selectivo, proporciona incertidumbres adecuadas y es de utilidad para cuantificar de manera rápida y certera los metales traza estudiados en matrices que puedan ser llevadas a solución acuosa, previa digestión en medio ácido.
Palabras clave: Validación; Manganeso; Molibdeno; Zinc; Plasma de acoplamiento inductivo.
Summary
A method to accurately determine and quantify manganese, molybdenum and zinc using Optical Emission Spectroscopy-Inductively Coupled Plasma by optical emission spectrometer was optimized and validated. Selectivity/ specificity, linearity, repeatability, intermediate precision and bias using reference materials and certified matrix reference material, respectively, were determined. Selected wavelengths (nm) Zn 213.857; Mn 257.610 and Mo 202.031 made it possible to discriminate spectral interferences. Linearity was proved by ad hoc statistical evaluations. Intermediate precision ranged between 5 and 11% and bias was never greater than 12%. Quantification limits (mg/L) were: Mn: 5; Zn: 10 y Mo: 10. Uncertainties associated to analytical determinations ranged between 10% and 16%. The validation of the proposed method demonstrated that it is selective, it provides adequate uncertainties and it is useful to quantify quickly and accurately the studied trace metals in matrices that can be taken intoaqueous solution with prior digestion in acid medium.
Key words: Validation; Manganese; Molybdenum; Zinc; Inductively Coupled Plasma.
Resumo
Descreve-se a validação de um método para a determinação de manganês, molibdênio e zinco a níveis-traço, em soluções aquosas, por Espectrometria de emissão por plasma acoplado indutivamente de argônio. Foi maximizada e validada a quantificação de manganês, molibdênio e zinco em solução aquosa ácida, utilizando um espectrômetro de emissão atômica por plasma acoplado indutivamente. Foram determinados: seletividade/especificidade, linearidade, repetibilidade e precisão intermediária usando materiais de referência, e viés contrastando contra material de referência certificado de matriz. Os comprimentos de onda (nm) selecionados foram: Zn 213.857; Mn 257.610 e Mo 202.031, os quais permitiram discriminar interferências espectrais. A linearidade das funções resposta foi provada por avaliações estatísticas ad hoc. A precisão intermediária variou entre 5 e 11% e o viés não ultrapassou 12%. Os limites de quantificação (μg/L) foram: Mn: 5; Zn: 10 e Mo: 10. As incertezas associadas com as determinações variaram entre 10 e 16%. A validação do método proposto demonstrou que é seletivo, proporciona incertezas adequadas e é útil para quantificar com rapidez e precisão os metais-traço estudados em matrizes que podem ser levadas a solução aquosa, prévia digestão em meio ácido.
Palavras-chave: Validação; Manganês; Molibdênio; Zinco; Plasma de acoplamento indutivo.
Introducción
La detección a nivel de trazas de elementos, tanto benéficos como tóxicos para seres humanos, animales, vegetales y medio ambiente, es necesaria en una amplia variedad de matrices (alimentarias, biológicas o ambientales) de gran importancia en el área de Salud Humana (1) (2).
Ese objetivo sólo puede lograrse mediante técnicas analíticas e instrumentales de elevada sensibilidad. Entre dichas técnicas, la espectrometría de emisión mediante plasma inductivo de argón (ICP-OES) es de gran sensibilidad y costo accesible. En este marco, la obtención de resultados exactos y confiables es imperativa, debido a la importancia de la presencia de concomitantes que podrían afectar la confiabilidad de los resultados (3)(4).
Manganeso, molibdeno y zinc son microelementos esenciales para el humano. Cada elemento tiene por lo menos un rol importante que cumplir dentro del organismo, y para cada uno hay un rango de ingesta dentro del cual se mantiene la homeostasis (5). Por lo tanto, la cuantificación es fundamental para optimizar las recomendaciones acerca de las cantidades de estos elementos que no producen deficiencias ni efectos adversos en el humano (6).
El creciente avance de la Química Analítica Instrumental ha permitido su detección a nivel de trazas, debido al importante aumento de la sensibilidad en las técnicas de última generación.
La obtención de resultados exactos y confiables es imprescindible en los laboratorios que realicen los ensayos pertinentes, para llevar a cabo estudios que ostenten la seriedad que este tema amerita. Para esto se debe implementar un sistema de calidad. En este marco, la validación de las metodologías de análisis de metales traza es un tema de fundamental importancia y es una práctica que los laboratorios deben realizar imperativamente si utilizan métodos propios (7)(8).
La Norma IRAM 301:2005 (9) y la guía provista por la norma ISO/IEC 17025:05, sección 5.4, proporcionan lineamientos para la ejecución del proceso completo de validación (10). Estas normas, perfectamente concebidas, mencionan estrategias a seguir para detectar si el proceso de validación fue llevado a cabo en forma certera y correcta (11).
Se realizó la validación de un método de ensayo para que la determinación de manganeso, molibdeno y zinc provea valores certeros y trazables, contemplando la obtención de sus parámetros típicos, que incluyen: especificidad (evaluación de interferencias), linealidad, rango de trabajo, límites de detección y cuantificación, precisión (repetibilidad y/o precisión intermedia), veracidad (expresada como sesgo), así como que tenga en cuenta el aseguramiento de la calidad de los resultados, el entrenamiento y la capacitación del personal.
Materiales y Métodos
REACTIVOS
Para todas las operaciones, incluidas la preparación de todos los estándares, reactivos y hasta el enjuague final del material de vidrio y de plástico, se utilizó agua destilada desionizada (<18,2 WW/cm, obtenida mediante un equipo Nano Pure (Barnstead, Boston, MA, EE.UU.). Se utilizaron reactivos calidad para análisis, salvo que se especificara otra calidad. Para preservar estándares y muestras se utilizó ácido nítrico concentrado ultra puro, diluido apropiadamente según el caso.
MATERIALES
Se utilizó material volumétrico calibrado para la preparación de los estándares y las soluciones de las muestras a analizar. La totalidad del material del laboratorio fue enjuagado, lavado con detergente no iónico, vuelto a enjuagar y sumergido durante 24 h en ácido nítrico al 10-15%. Posteriormente, fue enjuagado 3 veces con agua destilada y 3 veces con agua destilada desionizada.
MATERIALES DE REFERENCIA
Para la obtención de las funciones respuesta se utilizaron diluciones apropiadas de soluciones madres, marca ChemLab, de: manganeso (1000 mg/L), molibdeno (1000 mg/L) y zinc (1000 mg/L), todas ellas trazables a NIST o PTB. Para el control de calidad del método se utilizaron estándares marca Inorganic Ventures traceables a NIST. Todas las soluciones fueron almacenadas en botellas de polietileno, refrigeradas a 4 ºC. Las diluciones de trabajo se prepararon diariamente.
MATERIAL DE REFERENCIA CERTIFICADO DE MATRIZ
Se utilizó el material de referencia certificado - NIST SRM 1643e "Trace elements in water", preparado en una matriz acuosa acidificada con ácido nítrico aproximadamente 0,5% (v/v) debido a su semejanza con las soluciones a que deben ser llevadas las distintas matrices a estudiar.
EQUIPO
Para la detección de los analitos involucrados en este trabajo se utilizó un espectrómetro de emisión atómica de plasma inductivo de argón (OPTIMA 8000, Perkin Elmer), multielemental secuencial, con configuración axial/radial, provisto de dos detectores de estado sólido (Segmented Coupled Devices – SCD) que cubren un rango espectral de 163 a 782 nm. La resolución del sistema es 0,006 nm a 200 nm. Opera con un nebulizador de flujo cruzado asociado a una cámara de expansión Tipo Scott y un automuestrador (AS 90, Perkin Elmer). La antorcha, desmontable, está tallada en una sola pieza de cuarzo para el plasma y el gas auxiliar incluye un inyector de alúmina de 2,0 mm de diámetro, resistente a la corrosión de los ácidos, incluyendo fluorhídrico y agua regia. La muestra se introduce a través de una bomba peristáltica de tres canales controlada por un ordenador. El equipo es completamente operado por un software ad hoc, provisto por el fabricante (WIN LAB 32 Versión5.5).
MÉTODOS
Los parámetros instrumentales fueron optimizados contemplando aquellas variables críticas que influyen en la señal analítica, entre ellas: el modo de observación, que puede ser axial u horizontal, eligiéndose la modalidad axial para los elementos traza; los caudales de los gases de mantenimiento de la descarga, auxiliar y de nebulización, de cuya variación depende la relación señal/fondo utilizada para cuantificar a los analitos; la potencia incidente; los tiempos de lectura y las líneas espectrales. Las condiciones instrumentales relevantes utilizadas se detallan en la Tabla I.
Tabla I. Condiciones instrumentales

VALIDACIÓN DE LOS MÉTODOS DE ENSAYO
La validación incluyó la evaluación y determinación de (12):
• Selectividad/Especificidad: Se determinaron los analitos en presencia de posibles interferencias espectrales a bajo nivel y a concentraciones elevadas.
• Linealidad: Se utilizaron materiales de referencia certificados con trazabilidad a NIST para preparar disoluciones madres (de 1 y 10 μg mL-1), que dieron origen a "curvas de calibrado" de ocho estándares, considerando en la preparación las concentraciones de ácido nítrico (HNO3) de las disoluciones stock, de forma que todas ellas tuvieran idéntica matriz ácida (matrix matching). Se utilizó ytrio como estándar interno. Se usaron rangos dinámicos acotados para asegurar linealidad. Se optó por el uso de calibración externa por tratarse de soluciones ácidas diluídas y ausencia de interferencias interlementos.
• Repetibilidad y precisión intermedia: La precisión del método, expresada como repetibilidad, se evaluó mediante la realización de diez mediciones independientes de cada uno de los patrones utilizados en la "función respuesta" por cada analista, en intervalos cortos de tiempo. Una vez comprobada la consistencia de los resultados obtenidos por los analistas, se consideró que se los podía tratar como provenientes de un solo analista y se calculó la precisión intermedia (11).
• Sesgo: se evaluó contrastando los resultados obtenidos por el método desarrollado contra un material de referencia certificado de matriz de NIST.
• Límites de detección y cuantificación: Se evaluaron diez soluciones independientes de blancos fortificados. Los niveles de fortificación fueron seleccionados para obtener concentraciones de analito semejantes a las de los límites de detección (~0,010 mg analito/L). Se expresaron como tres veces el desvío estándar de la relación señal/ ruido a la longitud de onda analítica de cada elemento estudiado (3 s).
Los límites de cuantificación se consideraron al menos 10 veces la relación señal/ruido a la longitud de onda analítica de cada elemento estudiado (10 s).
Se utilizó material volumétrico calibrado para la evaluación de todos estos parámetros.
Resultados
La evaluación de los parámetros de validación se describe a continuación:
SELECTIVIDAD/ESPECIFICIDAD
La mayoría de las interferencias espectrales descritas en la bibliografía se pudieron minimizar utilizando las siguientes longitudes de onda (nm): Mo: 202.031; Zn: 213.857 y Mn: 257.610.
Las interferencias espectrales de la matriz fueron analizadas mediante el agregado de los distintos interferentes a disoluciones sintéticas que contenían los analitos de interés en diferentes concentraciones. Se observó que la línea de Mn podía llegar a presentar ligeras interferencias de Ru, Zr y Co; la de Mo con Os, Cr y Cd, y la de Zn con Ni, Hf, Fe y V. La resolución espectral del espectrómetro, estimada en unos 6 picometros para longitudes de ondas próximas a los 200 nm, permitiría discriminar la mayoría de las interferencias descritas. Asimismo, dejan de tener importancia debido a las intensidades que ostentan las líneas de los analitos, como puede verse en la Tabla II.
Tabla II. Posibles interferencias espectrales
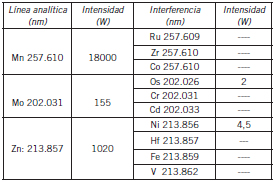
Sólo en el caso que las interferencias estuviesen presentes en concentraciones, en promedio, 50 veces mayores que los analitos a determinar, constituirían un problema en la medición de los elementos de interés de este trabajo.
Como se explicó más arriba, las líneas espectrales seleccionadas, se encuentran prácticamente libres de interferencias de la matriz e interferencias interelementos.
En las Figuras 1a, 1b y 1c se muestran las capturas de pantalla de los gráficos de las líneas utilizadas y la ausencia de interferencias espectrales importantes.
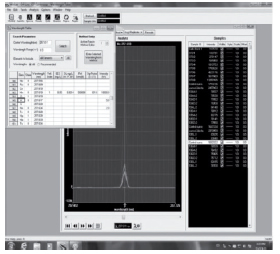
Figura 1 a. Captura de pantalla de líneas espectrales de manganeso.

Figura 1 b. Captura de pantalla de las líneas espectrales de molibdeno.

Figura 1 c. Captura de pantalla de las líneas espectrales de zinc.
LINEALIDAD
Para evaluar la linealidad se realizó una inspección visual de los valores obtenidos. Como aparentemente se trataba de regresiones lineales, se procedió a trazar la función respuesta ("curva de calibrado") que se ajustó por el método de cuadrados mínimos. El criterio de aceptación para el coeficiente de determinación se estableció en 0,995, lo cual implica que el modelo es explicativo en un 99,5% de los casos. El valor obtenido para estos coeficientes para los tres metales estudiados resultó siempre mayor que el valor impuesto.
En las Figuras 2, 3 y 4 se muestran las funciones respuesta obtenidas para los analitos estudiados, que provienen del promedio de los resultados obtenidos por los dos analistas que participaron de la validación.
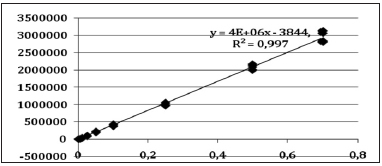
Figura 2. Funciones respuesta para manganeso.

Figura 3. Funciones respuesta para molibdeno.

Figura 4. Funciones respuesta para zinc
Se realizó adicionalmente el estudio de residuales y dos tests para asegurar la validez del ajuste. En la Tabla III se muestra el esquema de la salida del Programa ORIGIN 6.0 con los tests realizados para asegurar la validez del ajuste. El test t para contrastar la hipótesis de que la pendiente de la recta de ajuste es significativamente diferente de cero. En dicha Tabla III se observa que el valor de probabilidad para la pendiente (B) es <0,0001, lo cual significa que la probabilidad de rechazar la hipótesis de no diferencia de la pendiente con cero y equivocarse, es extremadamente pequeña. Por ello se rechaza la hipótesis de no diferencia y se acepta que la pendiente es significativamente diferente de cero. El nivel de significación es menor a 0,0001. En la misma tabla se muestran los resultados del análisis de la varianza (ANOVA). Aquí se prueba la hipótesis de si el modelo es significativo. Se compara si la variación de la variable dependiente, debida a la variación de la variable independiente, es superior a las variaciones introducidas por las variaciones propias de las variables. O sea, la suma de cuadrados explicada por el modelo es significativamente diferente de la suma de los cuadrados de los residuos. Por lo tanto, el hecho que la probabilidad de rechazar la hipótesis de no diferencia sea tan baja (<0,0001) hace que se pueda aceptar la diferencia significativa evaluada y el modelo es suficientemente explicativo.
Tabla III. Resultados de los tests t y F para asegurar la validez del ajuste para el Zn (salida del Programa ORIGIN 6.0).

Se muestra, a modo de ejemplo, en la Figura 5 el gráfico de residuales de Zinc.
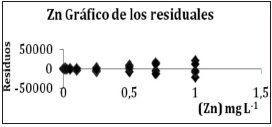
Figura 5. Residuales para el Zn.
PRECISIÓN: REPETIBILIDAD Y PRECISIÓN INTERMEDIA
Se evaluó la repetibilidad de cada analista y una vez comprobada la indistinguibilidad de los resultados obtenidos por ambos, se calculó la precisión intermedia del laboratorio a través de un análisis de varianza (ANOVA).
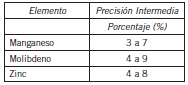
La precisión intermedia resultó ser del orden de la obtenida para técnicas que implican una puesta en solución de las muestras y su posterior determinación mediante métodos espectrométricos (Tabla IV).
Tabla IV. Resultados de la Precisión Intermedia de Mn, Mo y Zn.
VERACIDAD: SESGO DE LAS DETERMINACIONES
La veracidad del método, expresada como sesgo relativo porcentual, fue comprobada mediante el empleo del material de referencia certificado de matriz, NIST SRM 1643e "Trace elements in water". En este caso, cada analista realizó cinco mediciones independientes del material mencionado durante un tiempo equivalente a una jornada laboral. Para verificar la consistencia de los resultados obtenidos por los analistas también se llevó a cabo un análisis de varianza (ANOVA).
Los resultados obtenidos para el sesgo de las determinaciones resultaron bajos, lo que implicaría un reducido aporte a la incertidumbre de las mediciones, directamente relacionada con la exactitud del método de ensayo.
En la Tabla V se observan los valores certificados, su incertidumbre y el valor de cobertura para los analitos en estudio. Su incertidumbre está directamente relacionada con el sesgo y aporta a la incertidumbre del método validado. En la Tabla VI se observan los resultados obtenidos para cada analito y el sesgo porcentual obtenido.
Tabla V. Valores certificados, incertidumbre y valor de cobertura.

Tabla VI. Concentración hallada y sesgo

LÍMITES DE CUANTIFICACIÓN
En la Tabla VII se muestran los valores de los límites de detección y cuantificación obtenidos (en solución) para los elementos estudiados.
Tabla VII. Límites de detección y cuantificación

Discusión y Conclusiones
Los resultados del presente trabajo permiten asegurar el cumplimiento de requisitos normativos y legales impuestos para la determinación de Mn, Mo y Zn, en cantidades trazas, en muestras ambientales, biológicas y alimentarias, entre otras, con el equipo utilizado y en las condiciones del laboratorio en que fueron realizadas. Dichas muestras requieren como etapa preparativa su transformación en soluciones acuosas, que puede realizarse con mínimo riesgo de contaminación por digestión ácida. Ese paso tiene como objeto adecuar las muestras a los dispositivos de inyección, atomización y/o ionización en la descarga.
La dilución de las muestras favorece la disminución del efecto matriz en tal grado que permite el uso de una calibración externa del método de ensayo, lo que implica la preparación de los estándares de calibración en medio acuoso, ligeramente ácido. Este hecho hace posible la aplicación de este método validado a distintas matrices que pueden ponerse en solución en medio ácido.
El método propuesto, además de su rapidez, es de utilidad para cuantificar de manera certera manganeso, molibdeno y zinc con incertidumbres adecuadas, en distintas matrices que pueden ponerse en solución en medio ácido.
El método desarrollado ha sido perfectamente caracterizado en cuanto a su desempeño, y ha provisto niveles de precisión (expresada como precisión intermedia) y veracidad (expresada como sesgo) y límites de cuantificación compatibles con las necesidades planteadas para legislación medio ambiental, alimentaria, medicamentos y otras matrices de interés en el área de Salud humana.
AGRADECIMIENTOS
Este trabajo se ha realizado en el marco del Proyecto de Investigación aprobado por la Resolución 043/11 de la Carrera de Farmacia de la Facultad de Ciencias Exactas y Naturales de la Universidad de Belgrano, Ciudad Autónoma de Buenos Aires, Argentina.
Los autores agradecen la valiosa colaboración de la Comisión Nacional de Energía Atómica-CNEA, Buenos Aires, Argentina, de la Facultad de Farmacia y Bioquímica de la Universidad Nacional de Buenos Aires y de la Empresa Benito Roggio Ambiental SA, Ciudad Autónoma de Buenos Aires, Argentina.
1. WHO/FAO/ IAEA. Trace elements in human nutrition and Health World Health Organization. Geneva; 1996. [ Links ]
2. Mertz W. Review of the scientific basis for establishing the essentiality of trace elements. Biol Trace Element Res 1998; 66: 185-91. [ Links ]
3. Sandoval S. Validación de métodos y determinación de la incertidumbre de la medición: Aspectos generales sobre la validación de métodos. Guía Técnica No 1. Sección Metrología Ambiental y de Alimentos. Departamento de Salud Ambiental. Santiago de Chile; 2010. [ Links ]
4. Strathmann FG, Hoofnagle AN. Current and future applications of mass spectrometry to the clinical laboratory. Am J Clin Pathol 2011; 136: 609-16. [ Links ]
5. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Molybdenum, Nickel, Silicon, Vanadium and Zinc. Standing Committee on the Scientific Evaluation of Dietary References Intakes, Food and Nutrition Board & Institute of Medicine. National Academy of Sciences. Washington D.C.; 2001. [ Links ]
6. Horwitz W. Evaluation of analytical methods used for regulation of food and drugs. Anal Chem 1982; 54 (1): 67A- 76A. [ Links ]
7. EFSA (European Food Safety Authority)/FAO/OMS. Towards a harmonized TDS approach: a guidance document. EFSA J 2011; 9 (11): 2450. [ Links ]
8. The Fitness for Purpose of Analytical Methods. A Laboratory Guide to Method Validation and Related Topics. EURACHEM Guide. Editor: H. Holcombe. Teddington: LGC; 2014. [ Links ]
9. Norma IRAM 301:2005/ ISO/IEC 17025:2005. Requisitos generales para la competencia de los laboratorios de ensayo y de calibración. 4ta. Edición, Buenos Aires, Argentina. [citado 2005 mayo 9]. Consultado 25 de abril de 2016. Disponible en: http://www.chromu.com/calidad/DOCS/norma%20iso%2017025/iso_17025.pdf. [ Links ]
10. Criterios generales para la acreditación de laboratorios de ensayo y calibración según norma UNE-EN ISO/IEC 17025:2005 CGA-ENAC-LEC Rev. 6 Octubre 2014. [ Links ]
11. Butner J, Borth R, Boutwell JH, Broughton P MG. Approved Recommendation on Quality Control in Clinical Chemistry. Part 2. Assessment of Analytical Methods for Routine Use. Clin Chim Acta 1979; 98. F145-62. [ Links ]
12. Magnusson B, Näykk T, Hovind H, Krysell M. Handbook for calculation of measurement uncertainty in environmental laboratories. 3rd edition; Finland: Nordtest; 2012; 52; 2012. p. 52. [ Links ]
Recibido: 28 de abril de 2016
Aceptado: 10 de junio de 2016

