










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkActa bioquímica clínica latinoamericana
Print version ISSN 0325-2957On-line version ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.51 no.1 La Plata Mar. 2017
INMUNOLOGÍA
Mecanismo de exocitosis de gránulos citotóxicos: claves dadas a partir de la linfohistiocitosis hemofagocítica primaria
Exocytosis mechanism of cytotoxic granules: keys given from primary hemophagocytic lymphohistiocytosis
Mecanismo de exocitose de grânulos citotóxicos: Chaves dadas a partir de linfohistiocitose hemofagocítica primária
Silvia Leticia Monge-Rodríguez1a, María del Rosario Espinoza-Mora2b
1 MD.
2 PhD.
a Departamento de Fisiología, Departamento de Bioquímica, Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica.
b Departamento de Bioquímica, Escuela de Medicina, Universidad de Costa Rica, San José, Costa Rica. Hospital Calderón Guardia, Servicio de Medicina Interna, San José, Costa Rica.
CORRESPONDENCIA SILVIA LETICIA MONGE RODRÍGUEZ Dirección postal: 11501, Universidad de Costa Rica, Escuela de Medicina, Departamento de Fisiología, oficina 2-27. SAN JOSÉ – Costa Rica. Teléfono: 506 8400 8379 E-mail: silvia.monge.ro@gmail.com, leticia.monge@ucr.ac.cr
No hay conflictos de interés que declarar por ninguna de las autoras.
Resumen
La linfohistiocitosis hemofagocítica (HLH) es un síndrome clínico de hiperinflamación que se caracteriza por ser una respuesta inmune altamente estimulada pero inefectiva. En la HLH primaria se encuentran alterados el proceso de exocitosis de gránulos citotóxicos o los efectores que se encuentran en éstos, también existe afección de la activación de las células citotóxicas. Durante la exocitosis existe disfunción en la fase de transporte y maduración vesicular, en la regulación del proceso de docking y priming o en los complejos v-SNARE y t-SNARE. La conexión entre la célula citotóxica y célula diana se compromete si se afecta la proteína efectora perforina. SAP y XIAP se relacionan con la activación de las células inmunitarias. Aunque actualmente se conoce más de las moléculas que participan en la citotoxicidad, existe redundancia en las funciones de estas proteínas y aún quedan funciones que no han sido dilucidadas en dichos procesos.
Palabras clave: Linfohistiocitosis hemofagocítica; Exocitosis; Gránulos citotóxicos.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a clinical syndrome of hyperinflammation, in which the immune response is highly stimulated but it is ineffective. In primary HLH, the exocytosis process of cytotoxic granules or the effector proteins contained there are altered and the activation process of cytotoxic cells could be affected as well. During exocytosis there is dysfunction in vesicle maturation or translocation, in regulator proteins of the docking and priming process, or in v-SNARE and t-SNARE complexes. Connection between the cytotoxic cell and the target cell may be compromised if perforin effector protein is affected. SAP and XIAP have a role in the activation of immune cells. Though there is currently much known about the molecules participating in cytotoxicity, there is redundancy in protein functions involved in primary HLH, and there are some functions of these proteins that are still unknown.
Key words: Hemophagocytic lymphohistiocytosis; Exocitosis; Cytotoxic granules.
Resumo
A linfohistiocitose hemofagocítica (HLH) é uma síndrome clínica de hiperinflamação caracterizada por uma resposta imune que, apesar de ser altamente estimulada, é ineficaz. Na HLH primária, o processo de exocitose de grânulos citotóxicos, ou os efetores contidos neles, encontram-se alterados, também existe afecção na ativação das células citotóxicas. Existe disfunção na fase de transporte e amadurecimento vesicular, na regulação do processo de docking e priming, ou nos complexos v-SNARE e t-SNARE durante a exocitose. Caso a proteína efetora perforina estiver afetada, a conexão entre a célula citotóxica e a célula alvo está comprometida. SAP e XIAP estão relacionadas com a ativação das células imunitárias. Embora atualmente haja mais conhecimento a respeito das moléculas envolvidas na citotoxicidade, existe redundância nas funções destas proteínas. Contudo, ainda existem funções naqueles processos que não têm sido elucidadas até hoje.
Palavras-chaves: Linfohistiocitose hemofagocítica; Exocitose; Grânulos citotóxicos.
Introducción
Uno de los mecanismos por los cuales las células del sistema inmune median la citólisis es por la liberación de gránulos citotóxicos. La activación de linfocitos T CD8+ o de las células natural killer (NK) produce el acople de la maquinaria proteica implicada en la exocitosis, con lo cual se liberan los gránulos citotóxicos. Dentro de los componentes de los gránulos se encuentra la perforina, una molécula que ocasiona la interacción directa entre la célula citotóxica y la célula blanco, media la transferencia de granzimas a la célula blanco, además del choque osmótico e inducción de la muerte celular (1)(2).
La sinapsis inmunológica generalmente ocasiona un proceso de polarización de la célula citotóxica,. Esta corresponde a la interacción directa entre la célula presentadora de antígenos y la célula citotóxica (3) por medio de moléculas de membrana conocidas como complejo de activación supramolecular central (cSMAC, central supramolecular activation complex) y complejo de activación supramolecular periférico (pSMAC, peripheral supramolecular activation complex) (4).
La histiolinfocitosis hemofagocítica (HLH) es un síndrome clínico de hiperinflamación causado por la hipersecreción de citoquinas debido a una respuesta inmune muy estimulada pero que es inefectiva en el aclaramiento de patógenos. Las células que principalmente se ven hiperactivadas en este síndrome son los linfocitos T CD8+ y los macrófagos (2).
Los hallazgos clínicos claves en la HLH son fiebre, hepatoesplenomegalia y pancitopenia, además de datos de laboratorio característicos como ferritemia alta, transaminasas elevadas, hiperbilirrubinemia, hipertrigliceridemia, elevación de la cadena α del receptor de interleuquina 2 soluble (sCD25), y fibrinógeno en niveles bajos, en tanto que la hemofagocitosis como tal no siempre está presente (1)(2).
La etiología de HLH puede ser secundaria a malignidades (principalmente leucemias y linfomas), infecciones o procesos autoinmunes, o puede ser primaria debido a mutaciones en determinados genes que median la exocitosis de gránulos citotóxicos o la función de estos sobre la célula diana (2).
Las manifestaciones de HLH de etiología primaria se producen por detonantes como infecciones por gérmenes intracelulares tipo Candida sp., Histoplasma capsulatum o virus de Epstein-Barr. Lo anterior se debe a que en la HLH existe un compromiso de la respuesta inmune citotóxica, con lo cual no se elimina el antígeno o patógeno que induce la respuesta, por ende las señales inflamatorias persisten y se genera la hiperinflamación característica de HLH (5).
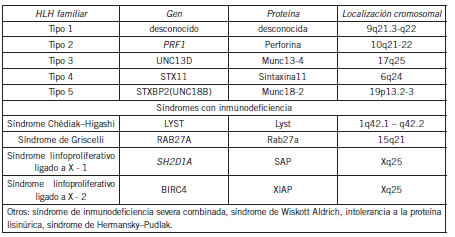
Los objetivos de esta revisión fueron describir las propiedades y funciones de las proteínas involucradas en la HLH primaria, establecer su papel dentro del mecanismo de exocitosis de gránulos citotóxicos en las cT CD8+ y las células NK e indicar su respectiva relación con los principales tipos de la HLH primaria (Tabla I) y las manifestaciones de este síndrome.
Tabla I. Clasificación de HLH primaria (1)(5).
El proceso de exocitosis
La exocitosis es el proceso que incluye el tráfico intracelular, el acople y la fusión de las vesículas con la membrana plasmática. Usualmente este mecanismo es mediado por distintos estímulos, aunque en la mayoría de las células la liberación de las vesículas es causada por el aumento intracelular de calcio (6)(7).
La exocitosis comprende distintas fases como la traslocación de la vesícula desde el aparato de Golgi hasta la membrana plasmática, la identificación de la zona activa en la membrana plasmática, el acople de la vesícula a la zona activa de la membrana (proceso conocido como docking), la preparación o activación de las vesículas (denominado priming), la fusión de la vesícula con la membrana celular y finalmente la liberación del contenido vesicular al medio extracelular (6). La exocitosis no es del todo difusional sino que se encuentra altamente regulada por distintos complejos proteicos (7).
AP y Lyst: proteínas envueltas en el tráfico vesicular
En el síndrome de Hermansky-Pudlak y el síndrome de Chédiak-Higashi se han identificado mutaciones en los genes que codifican para el complejo de la proteína adaptadora-3 (AP-3, adaptor protein complex 3) y la proteína reguladora del tráfico lisosomal (Lyst, lysosomal trafficking regulator protein) (8) (Tabla I). Ambas proteínas están involucradas en la traslocación de las vesículas desde los compartimentos intracelulares y en el acople al citoesqueleto (Figura 1) (8).
Las proteínas AP integran una familia de moléculas con 5 miembros identificados hasta el momento que median la endocitosis y la exocitosis. Los complejos AP se unen a señales citoplasmáticas principalmente en sitios con alta densidad de tirosina o de dileucina, reclutan clatrina y otras proteínas de tráfico vesicular (9).
Las AP son proteínas heterotetraméricas; algunas de las subunidades que las conforman son tejido-específicas principalmente neuronales, mientras que otras tienen distribución ubicua (10).
Las subunidades de los complejos AP tienen como estructura general un dominio central y dos dominios denominados “orejas”, que constituyen extensiones de las dos subunidades externas que conforman el dominio central; estos dominios tipo oreja se unen al dominio central por una estructura tipo picaporte (10).
Los dominios centrales y los dominios en oreja funcionan como andamio entre lípidos de membrana, dominios de señales intracelulares, maquinaria de fusión vesicular y otros componentes implicados en el ensamblaje vesicular (9). Por estudios con microscopía tridimensional y fluorescencia se ha visto un ensamblaje continuo de estos complejos AP. En zonas cercanas a la membrana plasmática predomina la AP-2 y participa en la endocitosis mediada por clatrina, mientras que en las zonas citoplasmáticas más cercanas al aparato de Golgi predominan la AP-1 y la AP-3 (11).
Todos los dominios de las AP tienen muchas interacciones, como sitios de fosforilación (principalmente en el dominio central) y de regulación (10). El factor de ribosilación–ADP (Arf, ADP-ribosylation factors) es una proteína GTP monomérica que interviene en el reclutamiento de las AP. La actividad GTPasa de Arf es baja por lo que depende de proteínas activadoras de Arf (ArfGAP, Arf GTPase activating proteins) (9).
Lyst es una proteína que pertenece a una familia de moléculas que contienen el dominio BEACH (BDCP, BEACH-domain containing proteins), denominado así por Beige y Chédiak–Higashi, respectivamente corresponden al modelo animal de ratones beige donde se ha estudiado la función de Lyst y el síndrome en humanos en el cual se identificó la mutación de esta proteína. Hasta el momento se han descrito 8 proteínas humanas de esta familia (12).
Se ha observado que la deficiencia de Lyst ocasiona formación de vesículas gigantes, las cuales son poco móviles y no son capaces de liberar el contenido de los gránulos. Diferentes autores concluyen que Lyst regula la función de otras proteínas que ensamblan el lisosoma (12). Investigaciones en modelos animales como Drosophila mauve han develado un papel de homólogos de Lyst en la maduración fagosomal (13).
En un estudio que comparó las células mononucleares de sangre periférica de pacientes con síndrome de Chédiak–Higashi que eran homocigotas para la mutación de LYST con las células mononucleares de controles, se observó: 1. que las células de personas con Chédiak-Higashi presentaban ensamblaje y fusión de endolisosomas normales, con lo cual se obtenían lisosomas aumentados de tamaño con diferentes centros densos, 2. que las proteínas de diferentes compartimentos endosomales se encontraban localizadas erróneamente en las organelas, 3. que los gránulos de las células T citotóxicas de estos pacientes se encontraban de forma excesiva anclados a la membrana, pero la fusión y liberación del contenido vesicular no se efectuaba aunque los gránulos tuvieran un tamaño normal y 4. que cuando se sobre-expresaba los efectores de exocitosis en las células T citotóxicas de estos pacientes se restablecía la dinámica de los gránulos secretores. Estos hallazgos demostraron que Lyst es necesaria en la maduración tardía de los gránulos y no solamente influye en el tamaño vesicular (14).
Papel de las proteínas GTPasas monoméricas
A las familias Rab y Rho de GTPasas monoméricas se les han descrito funciones facilitadoras o inhibidoras de las vías secretoras; estos mecanismos se han observado tanto en neuronas, células β pancreáticas o neutrófilos como en células citotóxicas (15–17).
En el síndrome de Griscelli tipo 2, en el cual se afectan principalmente los melanocitos y las células inmunes citotóxicas, se encuentra mutado el gen RAB27A (Tabla I) que codifica la proteína Rab27a, de la familia de GTPasasRab (Figura 1) (8)(15). Esta proteína tiene dentro de sus blancos a la proteína Munc 13-4 (Munc, acrónimo para mammalian uncoordinated), que regula el acoplamiento de la vesícula a la membrana, y está implicada en la estabilidad de la actina (15). Tanto en modelos animales como en humanos se ha visto que la polarización del gránulo necesaria durante la exocitosis no se afecta en estas células (18).
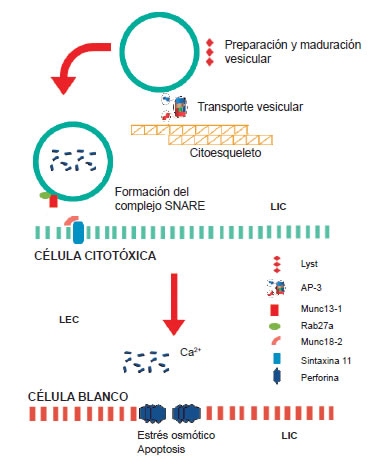
Figura 1. Proceso de exocitosis de gránulos citotóxicos y efecto sobre célula blanco. En la HLH primaria se han descrito mutaciones que causan afección del proceso de activación de la célula citotóxica (no se muestra en la imagen), del proceso de maduración y transporte vesicular como las proteínas Lyst y AP-3, en el docking y priming vesicular como las moléculas Munc13-4, Munc18-2 y sintaxina 11, o en la proteína efectora perforina, la cual mediante polimerización ocasiona un poro en la membrana de la célula blanco. En la parte superior de la imagen se representa un gránulo citotóxico que contiene a la perforina, la cual después del proceso de fusión en la membrana de la célula citotóxica (barras color verde) es liberada al medio extracelular y genera sus efectos sobre la membrana de la célula blanco (barras color café). Lyst: lysosomal trafficking regulator protein. AP-3: adaptor protein complex 3. Munc: mammalian uncoordinated, miembros de la familia Sec1/Munc18-like (SM). Rab: GTPasa monomérica de la subfamilia Rab de proteínas relacionadas con Ras. Sintaxina 11: proteína t-SNARE. Perforina: miembro de proteínas similares al complemento LEC: líquido extracelular, LIC: líquido intracelular. 128 Monge-Rodríguez SL y Espinoza-Mora M del R
Se ha observado en el modelo de ratón ashen, donde hay knockout del gen análogo de RAB27A, que la función de Rab27a es compensada por su isoforma Rab27b en algunas células como las plaquetas, pero que esto no sucede en melanocitos y células citotóxicas, ya que no expresan la isoforma Rab27b (19).
Acople de las vesículas a la zona activa de la membrana plasmática (docking) y preparación de las vesículas (priming)
Una vez la vesícula se ha traslocado cerca de las zonas activas en la membrana plasmática, debe ocurrir un proceso de acople de la vesícula con posterior acercamiento y activación (6). Esto ocurre por efecto de proteínas chaperonas de la formación del complejo SNARE (siglas por el nombre en inglés N-ethylmaleimide-sensitive factor attachment protein receptors), como son las moléculas de la familia Munc13 y Munc18 (miembros de la familia proteica Sec1/Munc18-like (SM), moléculas homólogas en mamíferos de proteínas unc de organismos como Caenorhabditis elegans (Figura 1) (16).
La familia de Munc13 tiene dos dominios de unión al calcio (C2), un dominio de unión a diacilglicerol (C1) y dos dominios homólogos Munc13 con los cuales interactúa con las proteínas SNARE (16)(20). Munc13-4 carece de dominio C1 y posee un dominio homólogo Munc13 [14]. Se ha constatado la expresión de Munc13-4 en muchas células hematopoyéticas y de isoformas de esta molécula en neuronas (21). Munc13-4 se encuentra mutada en HLH tipo 3 (Tabla I), por lo que los procesos de docking y priming se encuentran comprometidos (5).
En neutrófilos humanos, Munc13-4 rápidamente aumenta su traslocación a las membranas cercanas a las zonas activas, aproximadamente 30 segundos después de un estímulo como el aumento de la concentración de calcio intracelular, vuelve a los niveles basales 2 minutos después del estímulo (21). Algunas de las funciones que se le atribuyen a Munc13-4 es la disociación de Munc18-2 de sintaxina 11 y la promoción del ensamblaje del complejo SNARE (Figura 1) (16). Como ya se describió, la activación de Munc 13-4 por Rab27a/b es crucial para su función (15).
Fusión de la vesícula con la membrana plasmática
La interacción entre las proteínas v-SNARE (localizadas en la membrana de la vesícula) y t-SNARE (ubicadas en la membrana plasmática) es responsable de la fusión vesicular (22). En HLH tipo 5 se ha identificado una mutación en la región codificante de la proteína Munc18-2 (Tabla I), que es parte importante de ese proceso (5)(6).
Existen tres isoformas de Munc18 en mamíferos, Munc 18-1, Munc 18-2 y Munc 18-3, las cuales son parte de la familia Sec1/Munc18, cuyos miembros son proteínas altamente conservadas que participan en la fusión de la vesícula (23). Munc18-2 tiene estructura en forma de arco y sus dominios centrales interaccionan con la proteína sintaxina, que forma parte de t-SNARE (22). Munc18-2 en las plaquetas y en otras células inmunes se liga con sintaxina 11, pero también tiene afinidad por las sintaxinas 1, 2 y 3 en las células cromafines (16).
El complejo Munc18-2/t-SNARE es crítico para la estimulación de la fusión vesicular, ya que Munc18-2 constituye una plataforma para t-SNARE y facilita la estructura de v-SNARE/t-SNARE. Existe evidencia de que la fosforilación de Munc18-2 por la fosfolipasa C media aumento de la afinidad por la vesícula (24).
La sintaxina 11 pertenece a la familia de t-SNARE, y se relaciona con otra proteína de este complejo que es la SNAP-23. Estas proteínas se caracterizan por la presencia del dominio SNARE (25). La sintaxina 11 se afecta en la HLH de tipo 4 (Tabla I) (5)(6).
En análisis de células NK y cT CD8+ de pacientes con HLH tipo 4 se observó que las células que no expresaban Munc18-2 tampoco expresaban a la sintaxina 11. A partir de esto se dedujo que la integridad de la sintaxina 11 también ejerce regulación sobre Munc18-2 (22). Existe evidencia de que parte de la regulación de sintaxina 11 es por degradación de la proteína y que su adecuado funcionamiento depende de que la vía proteosómica se encuentre conservada. En células NK, la inhibición del proteosoma ocasiona disminución en la expresión de sintaxina 11 (25).
La perforina como molécula efectora
Posterior a la fusión del gránulo citotóxico a la membrana plasmática se da la liberación de distintas proteínas como granzimas y perforina(1). En la HLH tipo 2 se ha descrito la mutación del gen que codifica la perforina (Tabla I) (5)(6).
La perforina es una glucoproteína formada por varios dominios. En el extremo N-terminal tiene un sitio de unión al calcio, uno de los dominios centrales es hidrofóbico y le permite unirse a la membrana, otro de los dominios centrales tiene una región con abundantes residuos de cistina, homóloga a la del receptor de LDL. La molécula pertenece a proteínas similares al complemento (26).
La perforina se encuentra dentro de los gránulos en forma globular y en condiciones basales está inactiva por efecto de su interacción con la calreticulina. Su activación depende de la interacción con el calcio, con lo cual experimenta un cambio conformacional que le permite incorporarse a la membrana plasmática y polimerizarse (Figura 1). Aproximadamente 30 perforinas forman un poro de 14 nm de diámetro por donde las granzimas entran a la célula blanco y además se induce estrés osmótico (26).
Se ha documentado en cT CD8+ y en células NK de ratones y humanos que el fallo en la citotoxicidad mediada por granzimas y perforina ocasiona que la sinapsis inmunológica aumente su duración, mediando señales repetidas de calcio que conllevan a hipersecreción de citoquinas. El desacople entre estas células y la célula diana depende del inicio de señales de muerte celular (27).
SAP y XIAP
El síndrome linfoproliferativo ligado a X (XLP, X linked lymphoproliferative syndrome) se clasifica de acuerdo con las mutaciones asociadas (8)(28). En el tipo 1, la mutación está ubicada en el gen SH2D1A de la región Xq25, que codifica la proteína asociada con la molécula señalizadora de activación linfocítica (SAP, signaling lymphocytic activation molecule (SLAM) asociated protein) (28). El tipo 2 es causado por el gen XIAP/BIRC4, que se localiza de igual forma en Xq25 y consiste en seis exones que codifican la proteína inhibidora de apoptosis ligada a X (XIAP, X-linked inhibitor of apoptosis) (8)(28).
SAP es integrante de la familia de proteínas con dominios de homología Scr2 (SH2, Src homology domain 2). El dominio SH2 de SAP se une a motivos de tirosina en los dominios citoplasmáticos de inmunoreceptores SLAM, los cuales son altamente expresados en linfocitos B y T activados (29).
Los inmunorreceptores asociados a SLAM (como CD84 y Lys108) funcionan como intercambiadores de señales intercelulares entre células presentadoras de antígeno y células T o entre células T y células B (29)(30). SAP puede reclutar moléculas como la quinasa Fyn (proteína tirosina quinasa), la fosfatasa que contiene dominio inositol 1 (SHIP-1, SH2 domain containing inositol phosphatase) o la tirosina fosfatasa que contiene el dominio SH2 1 y 2 (SHP 1 y 2, SH2 domain containing tyrosine phosphatases 1 and 2), por lo que se postula que SAP propicia el cambio de una señal activadora o inhibidora (29).
Se han descrito dos principales formas de señalización de la vía SLAM–SAP–Fyn, a través de SHIP, con lo cual disminuye la producción de citoquinas, o la vía de la proteína quinasa C (PKC, protein kinase C), Bc110 y factor nuclear κB (NFκB, nuclear factor κB), que incrementa la producción de citoquinas como la interleuquina 4 (IL-4) (28). Células T con knockout de SAP exhiben dificultades en la producción de IL-4 después de la activación (31). Esta citoquina es clave en la polarización de la célula T hacia Th2 con generación de una respuesta inmune principalmente humoral (32).
XIAP pertenece a la familia de proteínas inhibidoras de apoptosis (IAP, inhibitor of apoptosis protein), que se caracteriza por la presencia del dominio de repetición baculovirus de IAP (BIR, baculovirus IAP repeats), el cual es un dominio de unión de residuos de zinc que fue descrito en baculovirus. XIAP contiene tres dominios BIR que pueden unirse a las caspasas 3, 7 y 9 e inhibir su función proteolítica (33)(34). XIAP se ha relacionado asimismo con la activación de vías de señalización celular como la relacionada con NFκB. No se ha logrado establecer con exactitud cómo se da la interacción entre XIAP y estas vías, pero se presume que la capacidad ligasa E3-ubiquitina de XIAP tiene un papel en esta función. Principalmente, las vías en que se propone un papel de XIAP es en la del receptor del factor de crecimiento transformante beta (TFG-β, transforming growth factor beta) y los receptores de reconocimiento de patrones moleculares (PRR, pattern recognition receptor) del sistema inmune innato (34).
Integración de la información sobre el mecanismo de exocitosis en HLH
En la HLH primaria se ha estudiado exhaustivamente la mayoría de los genes mutados involucrados y las proteínas que codifican. También existe un consenso general en cuanto a la correspondiente descripción fenotípica de cada uno de los tipos de HLH y del síndrome de inmunodeficiencia asociado. Sin embargo, aún quedan muchas dudas en cuanto a los mecanismos en los que están inmersas las proteínas afectadas y que se relacionan con la exocitosis de gránulos citotóxicos durante la respuesta inmunitaria efectora. Además, no son claras las funciones e interacciones que desempeñan dichas proteínas en otras líneas celulares no hematopoyéticas.
El fenotipo asociado a cada tipo de HLH depende de las células en donde se exprese la proteína en cuestión y si hay o no presencia de isoformas particulares en las células afectadas. Un ejemplo de este caso es el síndrome de Chédiak-Higashi, en donde aparte de inmunodeficiencia los pacientes presentan albinismo por afectación de los melanocitos (12).
La redundancia de los mecanismos celulares puede en ocasiones disminuir los efectos de la pérdida de la función proteica, con lo que se logra compensar total o parcialmente la deficiencia. Un ejemplo es lo que ocurre en ratones de estirpe ashen con algunas células que tienen función de Rab27b que sustituye a Rab27a, si bien es cierto en células citotóxicas no se da de esta forma por lo que la pérdida de Rab27a ocasiona de igual manera defectos en la exocitosis (19).
Algunas funciones de las proteínas afectadas en HLH fueron también observadas en otras líneas celulares como en las células β pancreáticas, en las neuronas y en las células cromafines. Entre estas líneas celulares existe homología de funciones proteicas como la observada en células citotóxicas. Es importante destacar que en realidad el mecanismo de exocitosis comparte muchos puntos en común entre células e incluso entre especies, por ejemplo la familia de Sec1/Munc18 se halla altamente conservada entre organismos como levaduras, Drosophila sp o mamíferos (23).
Usualmente cada proteína se asocia con una fase específica del proceso de exocitosis y por lo tanto con un determinado tipo de la HLH. Esta segmentación del proceso de exocitosis por etapas simplifica el proceso, ya que el paso entre traslocación – docking – priming – y la fusión vesicular depende de un continuo de funciones proteicas de la maquinaria involucrada, además, se postula que el docking y el priming constituyen un solo proceso (6). Munc13-4 y Munc18-2 son parte tanto del docking como del priming, están reguladas por proteínas G monoméricas, la diferencia clave entre ambas es que Munc13-4 se relaciona más con las proteínas v-SNARE y Munc 18-2 con t-SNARE.
El fallo en la citotoxicidad origina un síndrome de hiperinflamación como la HLH por la incapacidad de eliminar el antígeno que ocasiona la respuesta inmune citotóxica. Este antígeno genera continuamente estimulación del sistema inmune, con producción de citoquinas como el interferón gamma (IFN γ), que estimula los macrófagos y favorece el establecimiento de una respuesta T citotóxica. Parte de la efectividad del mecanismo citotóxico es también la terminación de la señal posterior al aclaramiento del antígeno (2). Se han realizado investigaciones que determinan no solo las proteínas involucradas, sino también la dinámica de la interacción entre la célula citotóxica y la célula blanco, que han proporcionado claves tanto sobre la fisiopatología de HLH como sobre los eventos moleculares que ocurren durante la sinapsis inmunológica y en la respuesta inmune posterior (27).
Las proteínas SAP y XIAP no forman parte del proceso de exocitosis como tal, pero están involucradas en señalización celular como integrantes de algunas vías importantes en la activación de células de la inmunidad innata como los linfocitos NK y de la inmunidad adaptativa como las células T, NKT y B. En los pacientes con síndrome de inmunodeficiencia ligado a X (Tabla I) se ha observado una disfunción de las células B, con baja producción de citoquinas relacionada posiblemente con alteraciones en las células B de memoria (28).
Pese a los remarcables avances en la comprensión de la fisiopatogenia del HLH, aún quedan bastantes preguntas por esclarecer. Muchas de las investigaciones se orientan a definir temporal y espacialmente en qué momento específico de la respuesta inmunológica aparecen en juego las proteínas comentadas previamente y de qué depende su activación. El enfoque de futuros estudios debe ser en definitiva dinámico, ya que una gran cantidad de estímulos, mecanismos regulatorios e inclusive el tipo de gérmenes causantes de la reacción inflamatoria pueden modificar la respuesta citotóxica.
Conclusiones
En la HLH primaria acontece una clara disrupción de los mecanismos de citotoxicidad. La exocitosis de gránulos citotóxicos se ve afectada por mutaciones en la maquinaria proteica participante, pero también se han descrito mutaciones asociadas con rutas de señalización molecular que se desencadenan previo a la exocitosis.
La hiperactivación inmunitaria característica de la HLH se debe al fallo en el aclaramiento de la respuesta y al fallo en la terminación de la respuesta. El fenotipo de la HLH dependerá del grado, célula afectada y proteína que se encuentre involucrada.
Con todo, persisten muchos puntos por definir en cuanto a las funciones de las proteínas implicadas en el mecanismo de exocitosis de gránulos citotóxicos, lo cual podría ser de gran relevancia para establecer nuevas dianas terapéuticas en pacientes portadores de HLH.
AGRADECIMIENTOS
Las autoras agradecen la colaboración y asesoría del Dr. Andrés Uribe, PhD y del Lic. Mauricio Parrales en la traducción del resumen del presente artículo al idioma portugués.
Referencias bibliográficas
1. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematol Am Soc Hematol Educ Progr 2013; 2013: 605–11.
2. Janka GE, Lehmberg K. Hemophagocytic syndromes - An update. Blood Rev 2014; 28 (4): 135–42.
3. Angus KL, Griffiths GM. Cell polarisation and the immunological synapse. Curr Opin Cell Biol 2013; 25 (1): 1–7.
4. Griffiths GM, Tsun A, Stinchcombe JC. The immunological synapse: a focal point for endocytosis and exocytosis. J Cell Biol 2010; 189 (3): 399–406.
5. Zhang L, Zhou J, Sokol L. Hereditary and Acquired Hemophagocytic Lymphohistiocytosis. Cancer Control 2014; 21(4): 301–12.
6. Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic 2008; 9 (9): 1414–24.
7. Dubuke ML, Maniatis S, Shaffer SA, Munson M. The Exocyst Subunit Sec6 Interacts with Assembled Exocytic SNARE Complexes. J Biol Chem 2015; 290 (47): 28245–56.
8. Brisse E, Wouters CH, Matthys P. Hemophagocytic lymphohistiocytosis (HLH): A heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev 2014; 26 (3): 263–80.
9. Park SY, Guo X. Adaptor protein complexes and intracellular transport. Biosci Rep 2014; 34 (4): 381–90.
10. Newell-Litwa K, Seong E, Burmeister M, Faundez V. Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci 2007; 120 (4): 531–41.
11. Kural C, Tacheva-Grigorova SK, Boulant S, Cocucci E, Baust T, Duarte D, et al. Dynamics of intracellular clathrin/AP1- and clathrin/AP3-containing carriers. Cell Rep 2012; 2 (5): 1111–9.
12. Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: A LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic 2013; 14 (7): 1199–216.
13. Mokhlasur R, Haberman A, Tracy C, Ray S, Kramen H. Drosophila mauve mutants reveal a role of LYST homologs late in the maturation of phagosomes and autophagomes. Traffic 2012;13 (12): 1680–92.
14. Sepulveda FE, Burgess A, Heiligenstein X, Goudin N, Menager MM, Romao M, et al. LYST Controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic 2015; 16: 191–203.
15. Moon TC, Befus AD, Kulka M. Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 2014; 5 (569): 1–18.
16. Fitch-Tewfik JL, Flaumenhaft R. Platelet granule exocytosis: a comparison with chromaffin cells. Front Endocrinol 2013; 4 (77): 1–11.
17. Kimura T, Niki I. Rab27a, actin and beta-cell endocytosis. Endocr J 2011; 58 (1): 1–6.
18. Krzewski K, Cullinane A. Evidence for defective Rab GTPase-dependent cargo traddic in immune disorders 2013; 18 (9): 1199–216.
19. Barral DC, Ramalho JS, Anders R, Hume AN, Knapton HJ, Tolmachova T, et al. Functional redundancy of Rab27 proteins and the pathogenesis of Griscelli syndrome. J Clin Invest 2002; 110 (2): 247–57.
20. Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry 2008; 47: 1474–81.
21. Pivot-Pajot C, Varoqueaux F, de Saint Basile G, Bourgoin SG. Munc13-4 Regulates Granule Secretion in Human Neutrophils. J Immunol 2008; 180 (10): 6786–97.
22. Côte M, Ménager MM, Burgess A, Mahlaoui N, Picard C, Schaffner C, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest 2009; 119 (12): 3765-73. [ Links ]
23. Xu H, Arnold MG, Kumar SV. Differential effects of Munc18s on multiple degranulation-relevant trans-SNARE complexes. PLoS One 2015; 10(9): 1–16.
24. Rodkey T, Liu S, Barry M, McNew J. Munc18a Scaffolds SNARE Assembly to Promote Membrane Fusion. Mol Biol Cell 2008; 19: 5422–34.
25. Dabrazhynetskaya A, Ma J, Guerreiro-Cacais AO, Arany Z, Rudd E, Henter J-I, et al. Syntaxin 11 marks a distinct intracellular compartment recruited to the immunological synapse of NK cells to colocalize with cytotoxic granules. J Cell Mol Med 2012; 16 (1): 129–41.
26. Osińska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol 2014; 1 (1): 109–15.
27. Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, Andrews DM, et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J Exp Med 2015; 212 (3): 307–17.
28. Yang X, Miyawaki T, Kanegane H. SAP and XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr Int 2012; 54 (4): 447–54.
29. Chu C, Wang Y, Zhang X, Ni X, Cao J, Xu W, et al. SAP-regulated T Cell-APC adhesion and ligation-dependent and -independent Ly108-CD3 interactions. J Immunol 2014; 193 (8): 3860–71.
30. Yan Q, Malashkevich VN, Fedorov A, Fedorov E, Cao E, Lary JW, et al. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci USA 2007; 104 (25): 10583–8.
31. Cannons JL, Wu JZ, Gomez-Rodriguez J, Zhang J, Dong B, Liu Y, et al. Biochemical and genetic evidence for a SAP-PKC-theta interaction contributing to IL-4 regulation. J Immunol 2010; 185 (5): 2819–27.
32. Allen JE, Wynn T. Evolution of Th2 immunity: A rapid repair response to tissue destructive pathogens. PLoS Pathog 2011; 7 (5): 5–8.
33. Hsieh WC, Chuang YT, Chiang IH, Hsu SC, Miaw SC, Lai MZ. Inability to resolve specific infection generates innate immunodeficiency syndrome in Xiap-/- mice. Blood 2014; 124 (18): 2847–57.
34. Galbán S, Duckett C. XIAP as ubiquitin ligase in cellular signaling. Cell Death Differ 2010; 17 (1): 54–60.
Recibido: 4 de enero de 2016
Aceptado: 28 de junio 2016

