










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkActa bioquímica clínica latinoamericana
Print version ISSN 0325-2957On-line version ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.51 no.3 La Plata Sept. 2017
HEMATOLOGÍA
Talasemias: Aspectos clínicos*
Thalassemias: Clinical aspects
Talassemias: Aspectos clínicos
Gustavo Chiappe1
1 Médico.
* Subcomisión de Eritropatías, Sociedad Argentina de Hematología, Ciudad Autónoma de Buenos Aires, Argentina.
CORRESPONDENCIA DR. GUSTAVO CHIAPPE Hospital Milstein. CABA, Argentina E-mail: gustavochiappe@gmail.com
Resumen
Los síndromes talasémicos, junto con las hemoglobinopatías talasémicas, las hemoglobinopatías estructurales y los síndromes de sobreexpresión representan las diferentes formas clínicas de las hemoglobinopatías. Los defectos genéticos responsables de los síndromes talasémicos determinan la síntesis disminuída o nula de la cadena de globina correspondiente. Según la cadena cuya síntesis es defectuosa, los síndromes talasémicos se clasifican en a-talasemias, b-talasemias, etc. Según las diferentes combinaciones de fenotipos particulares, las a-talasemias se clasifican en silente, portador, enfermedad con hemoglobina H e hidropesía fetal, y las b-talasemias en menor, intermedia y mayor. La sospecha diagnóstica de los síndromes talasémicos leves y de las hemoglobinopatías talasémicas es fácil a partir de la anemia leve con marcada microcitosis hipocrómica, ausencia indudable de ferropenia y cuadro familiar positivo. La electroforesis de hemoglobina con una cuantificación de hemoglobina A2 mayor de 3,5% prácticamente confirma el diagnóstico de una b-talasemia menor, mientras que una hemoglobina A2 normal o disminuida va a hacer sospechar una a-talasemia leve cuyo diagnóstico debe ser confirmado por estudio de ADN. Una vez establecida la condición talasémica del propósito es imprescindible identificar qué familiares consanguíneos son o no portadores del mismo gen talasémico, y estudiar a los cónyuges de los talasémicos detectados, a fin de prever, a través del consejo genético, el nacimiento de hijos homocigotas o doble heterocigotas con formas más severas de talasemias o hemoglobinopatías.
Palabras clave: Anemia; Talasemia; Hemoglobinopatía; Electroforesis de hemoglobina.
Abstract
Thalassemic syndromes, together with thalassemic hemoglobinopathies, structural hemoglobinopathies and over-expression syndromes represent the different clinical presentations of hemoglobinopathies, which are the mutational or deletional defects of globin genes. Genetic defects responsible for thalassemic syndromes determine a reduced or a lack of synthesis of the related chain. According to the defective synthesized chain, thalassemias are classified into a-thalassemias, b-thalassemias, etc. Depending on the different combinations of two or more phenotypes, a-thalassemias are classified into silent, carrier, Hb H disease and fetal hydrops, while b-thalassemias are classified into minor, intermediate and major b-thalassemia. Diagnostic suspicion of mild thalassemic syndromes and thalassemic hemoglobinopathies is easy based on a mild anemia with pronounced microcytosis and hypochromia, unquestionable absence of iron deficiency and positive family background. Hemoglobin electrophoresis with A2 hemoglobin level higher than 3.5% almost confirms a b-thalassemia minor, while a low or normal A2 hemoglobin level makes mild a-thalassemia suspicious and diagnosis must be confirmed by DNA study. Once the thalassemic condition of the propositus is confirmed, it is essential to identify which consanguineous relatives are or are not carriers of the same thalassemic gene, and then to study the couples of all already identified thalassemic relatives, in order to forecast, through genetic counselling, the birth of homozygous or double heterozygous children with more severe thalassemic or hemoglobinopathic conditions.
Keywords: Anemia; Thalassemia; Hemoglobinopathy; Hemoglobin electrophoresis.
Resumo
As síndromes talassêmicas, junto com as hemoglobinopatias talassêmicas, as hemoglobinopatias estruturais e as síndromes de sobre-expressão representam as diferentes formas clínicas das hemoglobinopatias. Os defeitos genéticos responsáveis pelas síndromes talassêmicas determinam a síntese diminuída ou nula da cadeia de globina correspondente. Segundo a cadeia cuja síntese é defeituosa, as síndromes talassêmicas são classificadas em a-talassemias, b-talassemias, etc. Conforme as diferentes combinações de fenótipos particulares, as a-talassemias são classificadas em silente, portador, doença com hemoglobina H e hidropesia fetal, e as b-talassemias em menor, intermediária e maior. A suspeita diagnóstica das síndromes talassêmicas leves e das hemoglobinopatias talassêmicas é fácil a partir da anemia leve com marcada microcitose hipocrômica, ausência induvidável de ferropenia e quadro familiar positivo. A eletroforese de hemoglobina com uma quantificação de hemoglobina A2 maior de 3,5% praticamente confirma o diagnóstico de uma b-talassemia menor, ao passo que uma hemoglobina A2 normal ou diminuída vai fazer suspeitar uma a-talassemia leve cujo diagnóstico deve ser confirmado por estudo de DNA. Assim que é estabelecida a condição talassêmica do propósito, é imprescindível identificar quais são os familiares consanguíneos e quais não são portadores do mesmo gene talassêmico, e estudar os cônjuges dos talassêmicos detectados, visando a prever, através do conselho genético, o nascimento de filhos homozigotas ou duplo-heterozigotas com formas mais severas de talassemias ou hemoglobinopatias.
Palavras-chave: Anemia; Talassemia; Hemoglobinopatia; Eletroforese de hemoglobina.
Introducción
La Genética es una de las ramas más jóvenes de la Medicina, que dio sus primeros pasos de la mano de las hemoglobinopatías y de los síndromes talasémicos. Tres “detalles” convergieron en este maridaje: a) HBB, HBA2 y HBA1 son genes pequeños (apenas 3 exones), b) su producto, la hemoglobina, es fácilmente “biopsiable” (apenas la punción de una vena o del pulpejo de un dedo) y c) la “célula” que aloja a la hemoglobina, el eritrocito, es el sitio de anidamiento del merozoíto palúdico. Frente a este azote de la humanidad todos los integrantes del glóbulo rojo se pusieron a la defensiva (mutaciones): hemoglobina (talasemias, drepanocitosis, enfermedad por Hb C), membrana (ovalocitosis del sudeste asiático, Duffy0) y enzimas (deficiencia de glucosa-6-fosfato deshidrogenasa). Pero estas mutaciones no son gratuitas: su costo es bajo en condiciones heterocigotas (ventajoso con respecto al costo palúdico) y alto en condiciones homocigotas o doble heterocigotas. Pero globalmente la ecuación “cierra”: ya lo había señalado John Haldane en 1948 (hipótesis palúdica) y de hecho en algunas zonas del sudeste asiático las talasemias están alcanzando frecuencias génicas de fijación. Los profesionales médicos deben lidiar a diario con estos males: ayudar a reconocer y aceptar el menor (talasemia menor, rasgo drepanocítico, etc.) y prevenir el mayor (talasemias intermedia y mayor, anemia drepanocítica, etc.).
La sospecha diagnóstica de los síndromes talasémicos leves y de las hemoglobinopatías talasémicas es fácil a partir de la anemia leve con marcada microcitosis hipocrómica, ausencia indudable de ferropenia y cuadro familiar positivo. La electroforesis de hemoglobina con cuantificación de las hemoglobinas A2 y Fetal permite una primera orientación diagnóstica: una hemoglobina A2 mayor de 3,5% prácticamente confirma el diagnóstico de una b-talasemia menor, mientras que una hemoglobina A2 normal o disminuida va a hacer sospechar una a-talasemia leve (o una db-talasemia si la hemoglobina Fetal está aumentada). La coexistencia de una ferropenia puede muchas veces dificultar el diagnóstico de un síndrome talasémico.
El estudio de ADN no es necesario para confirmar una b-talasemia menor típica, pero sí una b-talasemia menor atípica o dudosa, una b-talasemia intermedia o mayor, una a o una db-talasemia o una hemoglobinopatía talasémica. Una vez confirmada la condición talasémica del propósito es imprescindible identificar qué familiares consanguíneos son o no portadores del mismo gen talasémico, y estudiar a los cónyuges de los talasémicos detectados, a fin de prever, a través del consejo genético, el nacimiento de hijos doble heterocigotas con formas más severas de talasemias o hemoglobinopatías.
Estructura de la hemoglobina
La hemoglobina es la proteína eritrocitaria encargada del transporte del 100% del oxígeno desde los pulmones a los tejidos y del 25% del dióxido de carbono desde los tejidos a los pulmones. Estructuralmente es un heterotetrámero compuesto por dos cadenas de tipo alfa y dos cadenas de tipo no alfa (o, más precisamente, un homodímero en el que cada dímero está compuesto por una cadena de tipo alfa y una cadena de tipo no alfa). La estructura terciaria de cada cadena de globina determina la conformación de un bolsillo hidrofóbico en el que se aloja el grupo prostético hemo (protoporfirina IX + hierro). Los genes que codifican para las cadenas de globina de tipo alfa (de 141 aminoácidos) se localizan en 16p13.3 (de 5' a 3': z con expresión en etapa embrionaria y a2 y a1 en etapas fetal y adulta) y los que codifican para cadenas de globina de tipo no alfa (de 146 aminoácidos) en 11p15.5 (de 5' a 3': e con expresión en etapa embrionaria, Gg y Ag en etapa fetal y d y b en etapa adulta). Se forman entonces diferentes variantes normales de hemoglobina: Gower I (z2e2) en etapa embrionaria, Fetal (a2g2) en etapa fetal y A (a2b2) y A2 (a2d2) en etapa adulta (Fig. 1). En condiciones patológicas se pueden formar homotetrámeros como las hemoglobinas Bart's (g4) y H (b4). En el individuo adulto la mayor parte de la hemoglobina es A, representando la A2 menos del 3,5% y la Fetal menos del 2%.
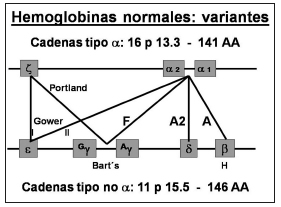
Figura 1. Variantes normales de hemoglobina en etapas embrionaria, fetal y adulta.
Hemoglobinopatías
DEFINICIÓN Y CLASIFICACIÓN
Las hemoglobinopatías resultan de defectos (mutacionales o delecionales) en la estructura de los genes que codifican para las cadenas de globina: HBA2, HBA1 (ambos codifican para cadenas a con idéntica estructura primaria), HBB, HBD, HBG1, HBG2 (los dos últimos codifican para cadenas g que difieren en un solo aminoácido: alanina y glicina respectivamente), etc. Dichos defectos determinan una alteración a) en la cantidad de globina sintetizada (defecto cuantitativo) y/o b) en su estructura primaria (defecto cualitativo), dando lugar a diferentes tipos de hemoglobinopatías (Fig. 2).
– Hemoglobinopatías estructurales (S, C, D, etc.): el defecto, generalmente mutacional, determina una alteración (con o sin implicancias funcionales) de la estructura primaria de la cadena de globina.
– Síndromes talasémicos (alfa, beta, delta/beta, etc.): el defecto determina la síntesis disminuída (en este caso con estructura primaria normal) o ausente de la cadena de globina afectada (1).
– Hemoglobinopatías talasémicas (Lepore, E, Constant Spring, etc.): el defecto (único) determina simultáneamente la síntesis disminuída de una cadena de globina con alteración de su estructura primaria.
– Síndromes de sobreexpresión (triple alfa, persistencia hereditaria de hemoglobina fetal, etc.): el defecto determina la síntesis en cantidad aumentada de una cadena de globina con estructura primaria normal.
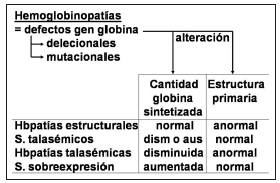
Figura 2. Diferentes cuadros clínicos de las hemoglobinopatías.
Para referirse a los diferentes genotipos que determinan estos defectos genéticos se usan los siguientes simbolismos: b (normal), bx (hemoglobinopatía estructural no identificada), bS (hemoglobinopatía S, ídem para las otras variantes estructurales: bC, bE, etc.), b0 (genotipo talasémico con síntesis nula de una cadena de globina), b+ (genotipo talasémico con síntesis disminuída de una cadena de globina estructuralmente normal), bX+ (hemoglobinopatía talasémica con síntesis disminuída de una cadena de hemoglobina estructuralmente anormal). La misma nomenclatura es válida para las otras variantes de hemoglobina, habitualmente refiriendo a los genes alfa duplicados como aa, mientras que axa significa un defecto estructural localizado en el gen HBA2, -a un genotipo a+ por defecto delecional del gen HBA2, etc.).
Síndromes talasémicos y hemoglobinopatías talasémicas
CONCEPTO Y CLASIFICACIÓN
El defecto genético va a determinar que a partir de ese gen defectuoso se sintetice una cantidad nula o disminuída de la cadena de globina correspondiente (2). En caso de que se sintetice una cantidad disminuída de globina, si ésta tiene una estructura primaria normal el cuadro es de un síndrome talasémico, en caso de que la estructura primaria esté, además, alterada el cuadro es de una hemoglobinopatía talasémica. Según el gen afectado las talasemias se clasifican en alfa-talasemia, beta-talasemia, delta/beta-talasemia (las más relevantes en adultos), gamma-talasemia, delta-talasemia, etc. (3)
Las alfa-talasemias suelen ser delecionales: genotipo a+ (-a o a-) cuando está delecionado uno de los dos genes alfa, a0 (--) cuando están delecionados ambos genes alfa (4). Las beta-talasemias suelen ser mutacionales: genotipo b+ o b0 según que el defecto determine la síntesis disminuída o nula de la cadena de globina b. La hemoglobinopatía talasémica más frecuente en nuestro medio (Lepore) es delecional (deleción de 7.4 Kb que incluye la parte distal -3'- del gen HBD y la parte proximal -5'- del gen HBB). El cuadro clínico común de todos los síndromes talasémicos y hemoglobinopatías talasémicas es una anemia microcítica hipocrómica de grado variable según el determinante más importante de severidad clínica, que es el disbalance entre la cantidad de cadenas de tipo alfa y de tipo no alfa sintetizadas (5).
Beta-talasemia
CLASIFICACIÓN
Hay tres cuadros clínicos de beta-talasemia: menor, intermedia y mayor (6). La menor es siempre heterocigota para genotipos b+ o b0. La talasemia intermedia puede ser homocigota para genotipos b+, doble heterocigota para genotipos b+ o b0 y triple alfa (lo más común en nuestro medio), rara vez heterocigota para genotipo b0 por mutaciones en el tercer exón (talasemias dominantes). La talasemia mayor es homocigota para genotipos b0 o doble heterocigota para genotipos b0 y b+. No es infrecuente encontrar pacientes con cuadros clínicos límites entre talasemia mayor e intermedia, entre intermedia y menor, o incluso entre talasemia menor y normal, difíciles de definir (7).
Talasemia menor
CLÍNICA
Se supone que entre un 1 y 2% de la población argentina es portadora de talasemia menor, en muchos casos sin saberlo. Habitualmente asintomática, depara una anemia microcítica hipocrómica leve, perfectamente sospechable a partir de un hemograma realizado con contador hematológico automático (8). A diferencia de la anemia ferropénica, en la que la disminución de los valores hematimétricos es paralela a la de los índices hematimétricos, en la talasemia menor es típica la disminución leve de los valores frente a una disminución significativa de los índices (situación semejante se puede encontrar en caso de una anemia ferropénica moderada/severa al cabo de unas 3-4 semanas de tratamiento adecuado con hierro, momento en el cual los valores hematimétricos ya están en franca mejoría pero los índices recién comienzan a corregirse) (9). Es importante recordar que las microcitosis (VCM<80 fL o HCM<27 pg) pueden ser hipocrómicas o no hipocrómicas. Mientras las microcitosis no hipocrómicas se deben generalmente a la presencia de esquistocitos (microangiopatías trombóticas, valvulopatías calcificadas, hemoglobinuria de la marcha, formas severas de membranopatías, etc.), las hipocrómicas resultan de una síntesis deficiente de hemoglobina. Puesto que la hemoglobina está compuesta por hem (protoporfirina IX y hierro) y globina, se puede deducir que las microcitosis hipocrómicas pueden deberse a defectos en la síntesis de protoporfirina IX (anemias sideroblásticas hereditarias, muy raras), deficiencia de hierro (la causa más frecuente de anemia) o defectos en la síntesis de globina (síndromes talasémicos y hemoglobinopatías talasémicas) (10). En la práctica es posible manejarse con el criterio de que toda anemia microcítica hipocrómica no ferropénica es muy probablemente talasémica.
Talasemia menor
DIAGNÓSTICO PROTEICO
Descartado fehacientemente el componente ferropénico como responsable de la anemia microcítica hipocrómica del paciente, corresponde investigar la posibilidad de que se trate de un cuadro talasémico, mediante la morfología eritrocitaria (microcitosis e hipocromía marcadas, ovalocitosis, dianocitos, punteado basófilo), el estudio familiar (la presencia de igual cuadro hematimétrico en algún familiar consanguíneo del propósito -uno de los padres o 50% de hermanos o hijos- es altamente orientador) y la electroforesis de hemoglobina (11). Habitualmente ésta muestra una concentración elevada (>3.5%) de hemoglobina A2, con hemoglobina Fetal normal, con lo que ya queda definido el cuadro (12). La presencia de una banda anómala en posición bastante semejante a la de S a pH alcalino y en una cuantificación entre 10 y 20%, sumada al cuadro talasémico con Hb A2 disminuída en un paciente de ascendencia mediterránea hace sospechar una Hb Lepore (Fig. 3), mientras que una banda anómala en posición de C a pH alcalino y una cuantificación de 20- 30% sumada al cuadro talasémico en un paciente de ascendencia oriental hace pensar en Hb E (13). Ambas hemoglobinopatías talasémicas deben confirmarse con estudio genético.
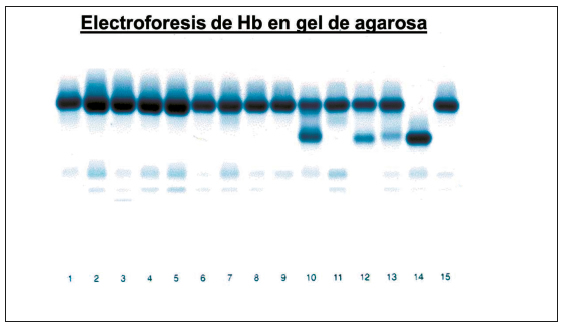
Figura 3. Electroforesis de hemoglobina en acetato de celulosa a pH 8.6
Calles 2, 4, 5, 7 y 11: talasemia menor - Calles 10 y 12: rasgo drepanocítico - Calle 13: Hb Lepore - Calle 14: S/beta0 talasemia
La presencia de punteado basófilo en el frotis de sangre periférica y de una hemoglobina A2 >3,5%, típicos de una talasemia menor, pueden no ser evidentes en caso de ferropenia concomitante, por lo que en este caso es importante corregir primero y en forma total la ferropenia para sólo entonces realizar el estudio hematimétrico y electroforético (14).
Talasemia menor
DIAGNÓSTICO GENÉTICO
No es necesario realizarlo para confirmar el diagnóstico en caso de un paciente con cuadro típico de talasemia menor (hematimetría y morfología eritrocitaria típicas, estudio familiar positivo, hemoglobina A2 >3,5%), pero sí si el cuadro es atípico o limítrofe (dudoso entre talasemia menor y normal o entre talasemia menor e intermedia) o si se trata de una hemoglobinopatía talasémica (15).
Talasemia mayor
El recambio de hemoglobina Fetal por hemoglobina A ocurre en los primeros meses de vida, por lo que el niño con talasemia mayor nace hematológicamente normal pero se va anemizando rápidamente en el primer año de vida y el diagnóstico siempre se hace antes de los dos. La anemia es severa con requerimiento transfusional permanente (16). El diagnóstico debe confirmarse con el estudio proteico (electroforesis de hemoglobina) y genético (ADN) de los padres, ya que ambos son necesariamente portadores de un gen talasémico. El tratamiento consiste en a) un régimen de hipertransfusión regular más o menos mensual con una hemoglobina diana entre 9 y 11 g/dL, b) tratamiento quelante para disminuir la sobrecarga de hierro y c) eventualmente trasplante de células madre hematopoyéticas.
Talasemia intermedia
Cuadro clínico intermedio entre talasemia menor y mayor, frecuentemente con límites poco definidos. Se diferencia de una talasemia menor por un mayor grado de anemia con requerimiento transfusional ocasional, presencia de eritroblastos en sangre periférica y de ciertas complicaciones: sobrecarga de hierro, eritropoyesis inefectiva y/o extramedular, etc. (17). Se diferencia de una talasemia mayor por un menor grado de anemia sin requerimiento transfusional permanente (18). Dado que la sobrecarga de hierro es debida fundamentalmente al incremento de la absorción intestinal (vía portal) es común que la ferritinemia subestime la sobrecarga real de hierro, debiéndose recurrir a la medición de la concentración hepática de hierro (LIC) por resonancia magnética (o por biopsia hepática) para su correcta evaluación (19). También es frecuente que estos pacientes cursen su niñez con un cuadro hematológico leve propio de una talasemia menor, y que sólo se haga evidente la mayor severidad hacia la adolescencia o primeros años de adultez. Este agravamiento progresivo lleva a que algunos pacientes con talasemia intermedia lleguen a comportarse en edades avanzadas casi como una talasemia mayor y ameriten un régimen transfusional regular con el consiguiente incremento del tratamiento quelante (20).
Alfa-talasemia
CLASIFICACIÓN
Hay cuatro cuadros clínicos de alfa-talasemia: silente, portador, enfermedad con hemoglobina H e hidropesía fetal (21). El silente es heterocigota para un genotipo a+. El portador es heterocigota para un genotipo a0 u homocigota para dos genotipos a+. La enfermedad con hemoglobina H es doble heterocigota para un genotipo a+ y un genotipo a0 y la hidropesía fetal es homocigota para dos genotipos a0. Dado que la alfa-talasemia es frecuentemente delecional, estos cuadros clínicos se correlacionan con la pérdida de 1, 2, 3 ó 4 genes de alfa globina (HBA2 y/o HBA1) respectivamente. Las formas mutacionales, menos frecuentes, son en general un poco más severas que las delecionales correspondientes porque el sistema proteolítico debe degradar no sólo las cadenas de b globina excedentes sino también las de a globina anómalas.
Alfa-talasemia
DIAGNÓSTICO PROTEICO Y GENÉTICO
En la alfa-talasemia silente los valores e índices eritrocíticos están habitualmente en el límite inferior de la normalidad, mientras que el portador de alfa-talasemia tiene valores e índices eritrocíticos semejantes a los de una beta-talasemia menor típica (22). En ambos casos lo que primero llama la atención, además de la hematimetría y morfología eritrocitaria, es el porcentaje de hemoglobina A2 próximo a o por debajo del límite inferior del rango de referencia (tener en cuenta que esta situación también puede darse en casos de anemia ferropénica), ya que las escasas cadenas a tienen más afinidad por las cadenas b que por las d. La confirmación diagnóstica depende del estudio genético, pero es fundamental solicitar dicho estudio orientándolo hacia la búsqueda de genotipos a+ o a0 heterocigotas o a+ homocigota, para lo cual es fundamental el estudio hematimétrico previo de ambos padres o, eventualmente, de otros familiares cosanguíneos (23).
La enfermedad con hemoglobina H suele ser equivalente a una beta-talasemia intermedia, también de severidad muy variable. La identificación de la hemoglobina H es difícil a través de la electroforesis de hemoglobina, pero fácil mediante la coloración supravital con azul brillante de cresilo de un frotis de sangre periférica y la observación de cuerpos de inclusión típicos (pelotas de golf) (24). El estudio familiar y el genético ulterior son también la única manera de confirmar el diagnóstico mediante la identificación de un genotipo a+ y de un genotipo a0 coexistentes en el propósito (25).
En la hidropesía fetal hay una ausencia total de cadenas a que reemplacen a las z, por lo que la anemización intraútero es severa, con la consiguiente muerte fetal en el último trimestre o poco después del parto. El estudio genético de ambos padres revela la existencia de genotipos a0 en ambos.
Otras talasemias
Las delta-talasemias no tienen importancia clínica y sólo se sospechan en ocasión de estudios de hemoglobina glicosilada. Las gamma-talasemias pueden tener alguna importancia en etapa fetal (es probable que pasen frecuentemente desapercibidas), pero no postparto. En etapa adulta la delta-beta talasemia se comporta clínicamente igual que una beta-talasemia menor, pero con hemoglobina A2 baja y una elevación leve de la hemoglobina Fetal. La confirmación diagnóstica es por estudio genético. La Fig. 4 muestra una orientación diagnóstica de los diferentes tipos de síndromes talasémicos menores a partir del volumen corpuscular medio (o hemoglobina corpuscular media) y de las concentraciones de Hb A2 y Fetal. Los síndromes de sobreexpresión no son cuadros talasémicos porque el disbalance entre la síntesis de cadenas alfa y no alfa es leve, con un excedente de un tipo de cadena sobre el otro que habitualmente no llega a deparar consecuencias clínicas ni disminución del VCM/HCM por sí solo.

Figura 4. Orientación diagnóstica de las formas menores de talasemia.
Condiciones doble heterocigotas para una hemoglobinopatía estructural y una talasemia
En las hemoglobinopatías talasémicas un único defecto genético (mutacional en las hemoglobinas E y Constant Spring, delecional en la hemoglobina Lepore) es responsable a la vez de la hemoglobinopatía estructural (alteración cualitativa de la estructura primaria) y de la talasemia (alteración cuantitativa a+ o b+). El defecto está en un solo alelo, y es diferente de la condición doble heterocigota para una hemoglobinopatía estructural y un síndrome talasémico, por ejemplo, una S/beta-talasemia, en la que ambos defectos están en trans, el hemoglobinopático en un alelo y el talasémico en el otro, por lo que ambos defectos, si corresponden al mismo tipo de gen (alfa o no alfa), van a presentarse disociados en los padres e hijos del propósito.
Cuando ambos defectos están en sendos alelos de un mismo gen el cuadro hemoglobinopático se agrava, por ejemplo en el caso de una S/beta-talasemia, porque las cadenas bS de un alelo se sintetizan en cantidad normal, mientras que las cadenas bA del otro alelo se sintetizan en cantidad disminuída (b+) o nula (b0), por lo que habrá una cantidad residual pequeña o nula de hemoglobina A respectivamente. En ambos casos la concentración de hemoglobina S será francamente superior al 45-48% habitual en condición heterocigota sin beta-talasemia concomitante. Es imposible confundir una anemia drepanocítica homocigota para bS de una S/b+ talasemia, por la presencia de microcitosis y de una pequeña cantidad de hemoglobina A en este caso, pero sólo la microcitosis y el estudio familiar, además del estudio genético, permiten diferenciar una condición homocigota para bS de una S/b0 talasemia (Fig. 3).
Lo contrario ocurre en los casos de co-herencia, por ejemplo, de una hemoglobinopatía de cadena b (bS) con una alfa-talasemia: las pocas cadenas alfa sintetizadas se van a unir preferencialmente a las cadenas bA antes que a las cadenas bS, por lo que, cuanto más severo sea el cuadro alfa-talasémico, menor será la concentración (y consecuentemente más leve el cuadro clínico drepanocítico) de la hemoglobina S.
Consejo genético
Arribar al diagnóstico de una talasemia en un paciente obliga a identificar a todos los familiares cosanguíneos que puedan compartir el gen patológico y, a continuación, estudiar a todos los cónyuges de talasémicos identificados para detectar alguna patología que, co-heredada con la talasemia de la pareja, pueda dar lugar a cuadros clínicos más severos (26).
El primer objetivo es relativamente fácil, porque implica identificar qué familiares cosanguíneos presentan un cuadro hematológico más o menos semejante al del propósito: a veces con un simple hemograma alcanza para confirmar a los portadores y descartar a los normales, pero siempre conviene hacer el estudio completo incluyendo perfil de hierro y electroforesis de hemoglobina.
Mucho más difícil es la evaluación de los cónyuges, porque aquí el problema pasa por detectar “algo” que pueda co-heredarse junto con la talasemia del propósito y determinar en los hijos un cuadro clínico de severidad mayor. Dentro de ese “algo” se incluyen todas las talasemias del mismo tipo de gen (alfa o no alfa), incluyendo las variantes subclínicas con valores e índices eritrocíticos en el límite (a veces hay que acceder al estudio genético para clarificar estas situaciones límite), cualquier hemoglobinopatía (recordar que no todas dan banda anómala en la electroforesis a pH alcalino), síndromes de sobreexpresión, como la triple alfa (aaa, clínicamente silente en el portador heterocigota y sólo identificable por estudio genético), etc. El diagnóstico de una talasemia de tipo de gen distinto (alfa o no alfa) en el cónyuge (por ejemplo, propósito con beta-talasemia y cónyuge con alfa-talasemia, o viceversa) no suele ser problema porque el disbalance entre cadenas alfa y no alfa disminuye y el cuadro clínico se alivia.
Si se encuentra en el cónyuge alguna patología que pueda potenciarse en severidad clínica en co-herencia con la del propósito, se procederá al consejo genético consecuente (27). Si el consejo involucra un eventual estudio genético prenatal o preimplante, el estudio genético de ambos padres debe estar perfectamente realizado y confirmado antes de iniciar cualquier búsqueda de embarazo.
Conclusiones
Toda microcitosis hipocrómica fehacientemente no ferropénica tiene que hacer sospechar la posibilidad de un síndrome talasémico, ya que las anemias sideroblásticas hereditarias son mucho menos frecuentes. La confirmación de una b-talasemia heterocigota es fácil a partir del cuadro hematimétrico y de la morfología eritrocitaria, de una hemoglobina A2 aumentada y cuadro familiar compatible, pero no son infrecuentes los cuadros límites en los que es difícil el diagnóstico diferencial con la normalidad o con una talasemia intermedia. Si, por el contrario, la hemoglobina A2 está normal o disminuída corresponde sospechar una a-talasemia (o una db-talasemia si la hemoglobina Fetal está aumentada). El estudio genético está indicado para la confirmación de una a-talasemia (a partir del estudio familiar tratar de orientar hacia genotipos a+ o a0), de una b-talasemia menor atípica o dudosa, de una b-talasemia intermedia o mayor, de una db-talasemia o de una hemoglobinopatía talasémica (Lepore, E, Constant Spring, etc.). Dicho de otra manera, la b-talasemia menor típica, al igual que la condición de portador heterocigota de hemoglobina S (banda típica + drepanoformación positiva), son las dos únicas hemoglobinopatías cuya confirmación no requiere necesariamente el estudio genético.
CONFLICTO DE INTERESES: El autor declara no tener conflicto de intereses.
1. Martin A, Thompson AA. Thalassemias. Pediatr Clin North Am 2013 Dec; 60(6): 1383-91. [ Links ]
2. Rund D. Thalassemia 2016: Modern medicine battles an ancient disease. Am J Hematol 2016 Jan; 91(1): 15-21. [ Links ]
3. Higgs DR, Engel JD. Stamatoyannopoulos G. Thalassaemia. Lancet 2012 Jan 28; 379(9813): 373-83. [ Links ]
4. Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med 2013 Jan 1; 3(1): a011718.
5. Nienhuis AW, Nathan DG. Pathophysiology and clinical manifestations of the β-thalassemias. Cold Spring Harb Perspect Med 2012 Dec 1; 2(12): a011726.
6. Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet 2012 Jan 28; 379(9813): 373-83. [ Links ]
7. Fucharoen S, Weatherall DJ. Progress toward the control and management of the Thalassemias. Hematol Oncol Clin North Am 2016 Apr; 30(2): 359-71. [ Links ]
8. Cao A, Galanello R. Beta-thalassemia. Genet Med 2010 Feb; 12(2): 61-76. [ Links ]
9. Denic S, Agarwal MM, Al Dabbagh B, El Essa A, Takala M, Showqi S, et al. Hemoglobin A2 lowered by iron deficiency and -thalassemia: should screening recommendation for β-thalassemia change? ISRN Hematol 2013; 2013: 858294.
10. Danjou F, Anni F, Galanello R. Beta-thalassemia: from genotype to phenotype. Haematologica 2011 Nov; 96 (11): 1573-5. [ Links ]
11. Peters M, Heijboer H, Smiers F, Giordano PC. Diagnosis and management of thalassaemia. BMJ 2012 Jan 25; 344: e228. [ Links ]
12. Cappellini M-D, Cohen A, Eleftheriou A, Piga A, Porter J, Taher A. Guidelines for the Clinical Management of Thalassaemia. 2nd Edition Strovolos; Nicosia Cyprus: Talassaemia International Federation; 2008. [ Links ]
13. Olivieri NF, Pakbaz Z, Vichinsky E. HbE/β-thalassemia: basis of marked clinical diversity. Hematol Oncol Clin North Am 2010 Dec; 24(6): 1055-70.
14. Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. Int J Lab Hematol 2016 May; 38 Suppl 1: 32-40. [ Links ]
15. Higgs DR, Gibbons RJ. The molecular basis of α-thalassemia: a model for understanding human molecular genetics. Hematol Oncol Clin North Am 2010 Dec; 24(6): 1033-54.
16. Cao A, Moi P, Galanello R. Recent advances in β-thalassemias. Pediatr Rep 2011 Jun 16; 3(2): e17.
17. Matta BN, Musallam KM, Maakaron JE, Koussa S, Taher AT. A killer revealed: 10-year experience with beta-thalassemia intermedia. Hematology 2014 Jun; 19(4): 196-8. [ Links ]
18. Vichinsky EP. Non-transfusion-dependent thalassemia and thalassemia intermedia: epidemiology, complications, and management. Curr Med Res Opin 2016; 32(1): 191-204. [ Links ]
19. Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica 2013 Jun; 98(6): 833-44. [ Links ]
20. Musallam KM, Taher AT, Rachmilewitz EA. β-Thalassemia Intermedia: A clinical pespective. Cold Spring Harb Perspect Med 2012 Jul; 2(7): a013482.
21. Vichinsky EP. Clinical manifestations of α-thalassemia. Cold Spring Harb Perspect Med 2013 May 1; 3(5): a011742.
22. Origa R, Moi P. Alpha-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1435/. (Fecha de acceso: 29 de diciembre de 2016). [ Links ]
23. Harteveld CL, Higgs DR. Alpha-thalassaemia Orphanet J Rare Dis 2010 May 28; 5: 13. [ Links ]
24. Farashi S, Najmabadi H. Diagnostic pitfalls of less well recognized HbH disease. Blood Cells Mol Dis 2015 Dec; 55(4): 387-95. [ Links ]
25. Galanello R, Cao A. Gene test review. Alpha-thalassemia. Genet Med 2011 Feb; 13(2): 83-8. [ Links ]
26. Weatherall DJ. Thalassaemia: the long road from the bedside through the laboratory to the community. Transfus Med 2011 Aug; 21(4): 218-23. [ Links ]
27. Cao A, Kan YW. The prevention of thalassemia. Cold Spring Harb Perspect Med 2013 Feb 1; 3(2): a011775. [ Links ]
Recibido: 25 de julio de 2016
Aceptado: 4 de abril de 2017

