










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.51 no.3 La Plata set. 2017
HEMATOLOGÍA
Anemia microcítica-hipocrómica: anemia ferropénica versus b talasemia menor*
Microcytic and hypochromic anemia: iron deficiency anemia versus b thalassemia trait
Anemia microcitica-hipocrômica: anemia ferropênica versus b talassemia menor
Mónica Teresita Felisa Aixalá1
1 Bioquímica. Doctora de la Universidad de Buenos Aires.
* Subcomisión de Eritropatías. Sociedad Argentina de Hematología, Ciudad Autónoma de Buenos Aires, Argentina.
CORRESPONDENCIA Bioq. MÓNICA AIXALÁ Laboratorio Aixalá-Blanco Hematología-Hemostasia Zuviría 214-1424 - CABA, Argentina Teléfono: 11 15 4578 0749 E-mail: aixalamonica@gmail.com
Resumen
Las anemias microcíticas hipocrómicas (m-H) presentan VCM<80 fL y HCM<27 pg. Son producto de la baja biodisponibilidad del hierro (Fe), o del defecto de la síntesis de globinas o del HEMO. La más frecuente es la anemia por deficiencia de hierro (ADH), seguida por las talasemias y las anemias de procesos crónicos. Menos frecuentes son aquellas por defectos en el HEMO o por causas genéticas del metabolismo del Fe. El objetivo del trabajo es revisar, por medio de parámetros de distinta complejidad, diferencias entre ADH y b talasemia heterocigota (b-Tal-het), las m-H de mayor prevalencia en nuestro medio. Los recuentos de eritrocitos y reticulocitos, hemoglobina, ferremia, ferritina, saturación de la transferrina, HbA2, porcentaje de alteraciones morfológicas son menores en la ADH. El VCM, el HCM, la ADE, los índices de microcitosis, transferrina, y los receptores solubles de transferrina son menores en b-Tal-het. El estrés oxidativo está aumentado en ambas patologías. En el análisis de estos parámetros se discute el grado de deficiencia de Fe y/o la mutación de b-Tal-het. Se aplica un algoritmo para m-H a partir del Fe sérico. Una vez descartadas las m-H más comunes, se debe investigar a-Tal-het, la cual se considera la causa de la mayoría de m-H inexplicadas.
Palabras clave: Microcitosis; Deficiencia de hierro; Volumen corpuscular medio; Amplitud de la dispersión eritrocitaria; Hipocromía; Talasemia; Índices de microcitosis; Receptor de transferrina; Electroforesis de hemoglobina; Estrés oxidativo.
Abstract
Microcytic hypochromic anemia (m-H) presents MCV<80 fL and MCH<27 pg. m-H can result from iron availability, defects in globin or HEMO synthesis. The most frequent m-H is iron deficiency anemia (IDA), followed by thalassemias and anemia chronic disease. Rare m-H are a consequence of HEME defects or iron metabolism genetic defects. The aim of this study is to review the differential diagnosis between IDA and b thalassemia trait (b thal trait), the most frequent in our environment. Results of laboratory tests are analysed. Erythrocytes, hemoglobin, reticulocytes, iron, ferritin, transferrin saturation, HbA2 and percentage of morphologic changes are lower in IDA compared with b Thal trait. MCV, MCH, RDW, microcytic index, transferrin and soluble transferrin receptor are higher in IDA compared with b Thal trait. Oxidative stress is increased in the two forms of microcytoses. Degree iron deficiency in IDA and b Thal trait mutation must be considered in the analysis of the parameters. A flowchart is proposed to evaluate m-H stemming from serum iron value. After excluding the most frequent causes of microcytic anemia, a thalassemia trait must be considered.
Keywords: Microcytosis; Iron deficiency; Mean corpuscular volume; Red blood cell distribution width; Hypochromia; Thalassemia; Microcytosis discriminant indices; Transferrin receptor; Hemoglobin electrophoresis; Oxidative stress.
Resumo
As anemias microcíticas hipocrômicas (m-H) apresentam VCM<80 fL e HCM<27 pg. São produto da baixa biodisponibilidade do ferro (Fe), ou do defeito da síntese de globinas ou do HEMO. A mais frequente é a anemia por deficiência de ferro (ADH), seguida pelas talassemias e as anemias de processos crônicos. Menos frequentes são aquelas por defeitos no HEMO ou por causas genéticas do metabolismo do Fe. O objetivo do trabalho é revisar, através de parâmetros de diversa complexidade, diferenças entre ADH e b talassemia heterocigota (b-Tal-het), as m-H de maior prevalência no nosso meio. As contagens de eritrócitos e reticulócitos, hemoglobina, ferremia, ferritina, saturação da transferrina, HbA2, percentagem de alterações morfológicas são menores em ADH. O VCM, o HCM, a ADE, os índices de microcitose, transferrina, receptores solúveis de transferrina são menores em b-Tal-het. O estresse oxidativo está aumentado em ambas as patologias. Na análise destes parâmetros é discutido o grau de deficiência de Fe e/ou a mutação de b-Tal-het. Aplica-se um algoritmo para m-H a partir do Fe sérico. Depois de serem descartadas as m-H mais comuns, deve investigar-se a-Tal-het, a qual é considerada a causa da maior parte de m-H inexplicadas.
Palavras-chave: Microcitose; Deficiência de ferro; Volume corpuscular médio; Amplitude da dispersão eritrocitária; Hipocromia; Talassemia; Índices de microcitose; Receptor de transferrina; Eletroforese de hemoglobina; Estresse oxidativo.
ABREVIATURAS
ADE= dispersión del tamaño celular, grado de anisocitosis
ADH= anemia por deficiencia de hierro
a-Tal-het= a-talasemia heterocigota o menor
Anemia m-H= anemia microcítica hipocrómica
APC= anemia de los procesos crónicos
b-Tal-het= b-Talasemia heterocigota o menor
BMP= proteínas morfogenéticas
CHCM= concentración de hemoglobina corpuscular media
Dcytb= citocromo b duodenal
DMT1= transportador de metales divalentes
Fe= hierro
FNT= factor de necrosis tumoral
FPN= ferroportina
Ft= ferritina
G6PD= glucosa 6 fosfato deshidrogenasa
GR= recuento de glóbulos rojos o eritrocitos
Hb = Hemoglobina
HCM= hemoglobina corpuscular media
HFE= Proteína reguladora del Hierro en Hemocromatosls
HJV= hemojuvelina
Hp= hepcidina
Hto= hematocrito
IL-1= interleuquina 1
IL-6= interleuquina 6
INFg= interferón g
IPR= proteínas reguladoras del hierro
IRIDA= anemia por deficiencia de hierro refractaria al hierro
m-H= microcitosis-hipocromía
OMS= Organización Mundial de la Salud
SMADs= sma and mothers against decapentaplegic homologue
STEAP3= seis antígeno epitelial de membrana de la proteína de la próstata 3
sTfR= receptor soluble de transferrina
Tf= transferrina
VCM= Volumen corpuscular medio
Introducción
Anemia es el descenso de la hemoglobina total corporal y tiene una prevalencia mundial del 25%. Los niveles normales de hemoglobina (Hb) varían según edad, raza, sexo, y en determinadas situaciones como embarazo, altitud, tabaquismo, entre otras (1).
Hay diferentes clasificaciones de anemia. Las más usadas son la fisiopatológica y la morfológica.
En la clasificación fisiopatológica, además del hemograma, se evalúan los reticulocitos. Los reticulocitos, estadío anterior del eritrocito maduro, han perdido el núcleo de los eritroblastos y conservan el ARN, lo cual permite su identificación en el laboratorio. Así, la anemia podrá ser clasificada como regenerativa, si los reticulocitos están aumentados, y como arregenerativa, en caso contrario.
La clasificación morfológica se basa en el tamaño celular, a través de la medición del volumen corpuscular medio (VCM). De acuerdo con su valor, las anemias pueden ser clasificadas como normocíticas, macrocíticas o microcíticas.
En las anemias normocíticas, generalmente normocrómicas, el VCM varía entre 80 y 100 fL. Si bien no siempre es sencillo identificar una patología, se pueden necesitar otras pruebas de laboratorio. Entre ellas, el recuento de reticulocitos puede ayudar a establecer un diagnóstico diferencial (2). Si los reticulocitos están aumentados, se sospecha alguna hemoglobinopatía o de determinado momento post hemorragia o post crisis hemolítica. Si los reticulocitos están normales o disminuidos, se sospecha una menor producción de eritropoyetina (insuficiencias renal o hepática), o bien, de un estímulo disminuido o una menor respuesta de la médula ósea. Si el estímulo está disminuido, hay que considerar posibles endocrinopatías o desórdenes crónicos o malnutrición proteica, y si la respuesta es menor, hay que pensar en aplasia medular o mieloma.
Las anemias macrocíticas (VCM mayor de 100 fL) pueden ser megaloblásticas (fundamentalmente por deficiencia de vitamina B12 y/o folatos) o no megaloblásticas (hemólisis, alcoholismo, insuficiencia hepática, u otras por mecanismos poco conocidos) (3).
En las anemias microcíticas, generalmente hipocrómicas (anemia m-H), el VCM es menor de 80 fL y la hemoglobina corpuscular media (HCM) menor de 27 pg/dL. Las anemias m-H son las más comúnmente encontradas en la práctica médica. La deficiencia nutricional de hierro (Fe) y los rasgos talasémicos son las principales causas de estas anemias en pediatría (4).
El objetivo de este trabajo fue evaluar y establecer un diagnóstico diferencial entre las dos anemias m-H de mayor prevalencia en nuestro medio: anemia por deficiencia de hierro (ADH) y b-talasemia menor o heterocigota (b-Tal-het), con una previa referencia a otras anemias m-H.
Anemias microcíticas hipocrómicas: generalidades
Las anemias m-H son un grupo heterogéneo de patologías que pueden ser hereditarias o adquiridas. Son consecuencia de la baja biodisponibilidad del Fe, o de un defecto en las síntesis de globinas o del Hemo.
La eritropoyesis necesita una efectiva hemoglobinogénesis. La síntesis del Hemo precede a la síntesis de las globinas y activa la transcripción y traslación de las mismas (5). Durante la diferenciación y maduración del eritrocito, las globinas, el Hemo y el Fe deben estar en cantidades adecuadas para formar una Hb estable.
Las anemias m-H más comunes son la ADH, las talasemias (Tal) y algunas anemias sideroblásticas (Figura 1). Las anemias de los procesos crónicos (APC) generalmente son normocíticas y, en determinadas circunstancias, pueden ser microcíticas (Figura 2).
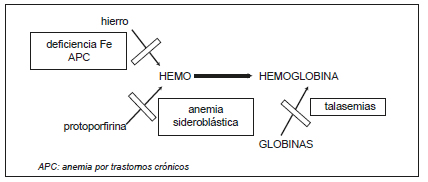
Figura 1. Alteración de la hemoglobinogénesis.
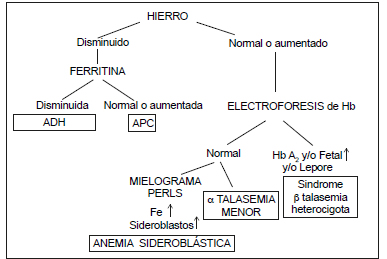
Figura 2. Algoritmo para anemias microcíticas a partir de la ferremia.
ADH: anemia por deficiencia de Fe. APC: anemia de los procesos crónicos.
Es importante conocer la causa de una anemia m-H y establecer un diagnóstico diferencial, a través de “estrategias” con distintos grados de complejidad.
Anemia por deficiencia de hierro (ADH)
La deficiencia de Fe pasa por diferentes estadíos: prelatente, latente y anemia manifiesta.
En el primero de los estadíos, hay descenso de la ferritina (Ft) plasmática y del Fe de depósito (con hemosiderina disminuida o ausente). En el estadío latente (eritropoyesis ferropénica), comienzan a aumentar la transferrina (Tf) y los receptores de transferrina (TfR) debido a la necesidad del eritroblasto de incorporar Fe para la síntesis de Hb. Tanto en la etapa prelatente como en la latente, no hay anemia.
En el tercer estadío hay anemia (por disminución de la Hb), con descenso de VCM y HCM (m-H). Es la llamada ADH. De acuerdo con la OMS (Organización Mundial de la Salud) y UNICEF (Fondo de las Naciones Unidas para la Infancia), la deficiencia de Fe es la principal causa de carencia nutricional (6).
La deficiencia de Fe es producto de la alteración del equilibrio entre las necesidades del individuo, y el aporte y/o la pérdida de Fe. Es importante no solamente la cantidad de Fe que contienen los alimentos sino también, su biodisponibilidad. El Fe que está dentro del grupo Hemo es el que se absorbe con mayor efectividad.
El Fe no-hemínico contenido en los alimentos se absorbe en menor porcentaje que el Fe-hemínico. La mayor parte del Fe de la dieta es de tipo no-hemínico y se encuentra en estado férrico. Este se reduce por la reductasa Dcytb (ferrirreductasa citocromo B duodenal) e ingresa por la membrana apical del enterocito a través de la DMT1 (transportador de metales divalentes), principal importador celular de Fe.
El Fe ingresado al enterocito, si no se usa para la síntesis de Ft, pasa a la membrana basolateral. Allí, la hefastina oxida al Fe y la ferroportina (FPN) lo exporta al plasma, donde es tomado por la Tf. Esta lo lleva a diferentes sitios y, fundamentalmente, a la médula ósea. El Fe se incorpora a los precursores eritroides, para la síntesis de Hb, por medio de los TfR. El Fe no utilizado se deposita como Ft y/o hemosiderina.
El Fe se elimina, principalmente, por vía intestinal, renal y por descamación epitelial, pero no hay ningún mecanismo regulatorio de excreción del Fe del organismo.
Hay un sistema de proteínas reguladoras del hierro (IRP), las cuales son “sensoras” del nivel de Fe. De acuerdo con éste, las IRP van a regular el aumento o disminución de la síntesis de diferentes proteínas (DMT1, TfR, Ft, entre otras) para la mejor absorción, utilización y/o depósito del Fe.
A medida que se acentúa la deficiencia de Fe van variando las concentraciones de las diferentes proteínas. Hay descenso de Ft y aumentos de DMT1 y TfR. Esto no sólo permite evaluar los estados de deficiencia de Fe sino también colaborar en el diagnóstico diferencial con otros desórdenes del metabolismo del Fe.
La deficiencia de Fe afecta especialmente a determinados grupos de riesgo, en los cuales es mayor el requerimiento de este nutriente (embarazadas, mujeres en edad fértil, preescolares, niños en el primer año de vida, adolescentes). El 50% de los casos de anemia en el mundo son por deficiencia de Fe y, por cada dos personas con deficiencia de Fe (estados prelatente y latente), hay una con anemia ferropénica manifiesta.
En Argentina, según datos de la Encuesta Nacional de Nutrición y Salud, la prevalencia de anemia ferropénica es del 34% en niños menores de 2 años (7) y del 20% en las mujeres en edad fértil. Esta prevalencia varía en las distintas zonas geográficas. Por ejemplo, en el Nordeste es del 45,7 y 22,6%, respectivamente, en estos dos grupos (8). En el mundo, alrededor de 468 millones de mujeres en edad fértil sufren ADH (9).
Anemia de los procesos crónicos
La APC es una de las causas más frecuentes de anemia en la práctica clínica y puede acompañar a infecciones, inflamaciones y neoplasias (10).
Su etiopatogenia responde a cuatro mecanismos: alteración en la utilización del Fe, disminución de la vida media de los eritrocitos, inhibición de la hematopoyesis y deficiencia relativa de eritropoyetina. Estas alteraciones se deben en gran medida a los efectos generados por el aumento de interferón g (INF-g), interleuquina 1(IL1), interleuquina 6 (IL6), factor de necrosis tumoral (FNT), que inhiben la eritropoyesis y afectan la homeostasis del Fe, induciendo la producción de hepcidina.
En las APC existe una incorporación insuficiente de Fe a los precursores eritroides para la síntesis de Hb, a pesar de existir una adecuada cantidad del Fe de depósito. Se trata de una deficiencia funcional de Fe (11) (12), o sea, que hay descenso de Fe plasmático, a pesar del incremento del Fe de depósito.
En las APC, la hepcidina (Hp) juega un rol central en su fisiopatología y en la regulación de la homeostasis del Fe. La Hp, secretada por el hígado, es un péptido de 25 aminoácidos, producto del gen HAMP (19q13.12) (13) (14). El aumento de Hp trae como consecuencia anemia y su descenso produce sobrecarga de Fe (15) (16).
Hay reguladores positivos y negativos de la síntesis de Hp. El incremento de Hp está regulado por IL6, infecciones, aumento de Fe, y otras proteínas del metabolismo del Fe (17).
La vía regulatoria, en la que participan proteínas morfogenéticas (BMP) y SMADs (sma and mothers against decapentaplegic homologue), posee una función central en la regulación de la síntesis de Hp que “responde” a los niveles de Fe en el organismo (por los aumentos de Tf diférrica o del nivel de Fe tisular). Proteínas como la hemojuvelina (HJV), TfR2, proteína reguladora del hierro en hemocromatosis (HFE) se relacionan, también con esta vía regulatoria, como sensores de la Tf diférrica. En procesos inflamatorios y/o infecciones hay aumento de Hp (con descenso del Fe plasmático) como consecuencia de su aumento transcripcional por medio de la IL6.
La hipoxia y la eritropoyesis son reguladores negativos de la síntesis de Hp (17) (18).
La FPN es el exportador de Fe de la célula (enterocito, hepatocito, macrófago). La Hp, al unirse a la FPN, induce la internalización y degradación del complejo Hp-FPN, generando la pérdida de la expresión funcional de la FPT. Cuando hay aumento tisular de Fe, aumenta la producción de Hp. Esta, en su interacción con la FPN, limita la absorción intestinal del Fe y la liberación del mismo de los macrófagos.
La APC es leve a moderada, generalmente normocítica pero, en determinadas circunstancias, puede ser microcítica. Cursa con Fe plasmático y Tf disminuidos e Fe de depósito aumentado.
Para la caracterización de esta anemia se puede medir el incremento de marcadores bioquímicos como la eritrosedimentación, proteína C reactiva, Hp e IL6.
Alteraciones de la hemoglobina
Las hemoglobinopatías son alteraciones hereditarias, con carácter autosómico recesivo. Se clasifican en hemoglobinopatías estructurales, talasemias, hemoglobinopatías talasémicas y síndromes de sobreexpresión.
La frecuencia de hemoglobinopatías en la población mundial es muy elevada y su distribución geográfica es variable. Aproximadamente un 5% de la población mundial es portadora de un gen de la Tal o de la HbS. El porcentaje de portadores Tal puede alcanzar el 25% en algunas regiones. Las Tal tienen una frecuencia variable según las zonas geográficas. Originalmente estaban limitadas a las regiones tropicales y subtropicales (coincidente con el área de la malaria), pero por las corrientes migratorias se han extendido a otras zonas (19-21).
Los síndromes Tal (thalassa: mar, hamia: sangre) son los trastornos genéticos (monogénicos) más prevalentes en el mundo, caracterizados por deficiencias variables de la producción de alguna de las cadenas de globina que la forman y con alteración de la proporción de las hemoglobinas normales (22). En las a-Tal hay deficiencia o ausencia en la síntesis de cadenas de globina y, en las b-Tal, hay deficiencia o ausencia en la síntesis de cadenas de b globina.
De acuerdo con el tipo y número de cadenas afectadas van a existir diferentes genotipos y fenotipos.
Hay cuatro genes a. Si hay deleción de uno de los genes (portador silencioso), generalmente no hay cambios ni en la hematimetría ni en la morfología. Cuando están afectados dos genes (a-Tal-het o a-Tal menor), hay microcitosis e hipocromía, además de otras posibles alteraciones morfológicas. Si se afectan tres genes, “aparece” una nueva Hb, la Hb H (tetrámero b). En estos pacientes hay hemólisis e importantes cambios del laboratorio hematológico. Si están afectados los cuatro genes (hidropesía fetal), hay muerte fetal o neonatal (Tabla I).
Tabla I. a talasemia.

Hay dos genes b. En la b-Tal-het está afectado un solo gen, y hay microcitosis e hipocromía. En la b-Tal homocigota están comprometidos los dos genes. En el homocigota, el VCM es mayor que en el heterocigota, y hay alta dispersión del tamaño celular (ADE). Esto se debe a la existencia de otros mecanismos adicionales como la eritropoyesis acelerada, compensadora de la hemólisis.
Hay además otros dos fenotipos b-Tal (talasemia mínima e intermedia) (Tabla II).
Tabla II. b talasemia.

Mientras que en las b+ talasemias hay reducción de la síntesis de cadenas b, en las b0 talasemias no hay síntesis de las mismas. Las de mayor porcentaje en nuestro medio son la mutación CD39 (b0) y la IVS-I-nt110 (b+), ambas prevalentes en la zona del Mediterráneo.
Dentro de los síndromes b talasémicos, aparte de la b- Tal-het, se encuentran la bd-Tal y la Hb Lepore. En el estado heterocigota, bd-Tal y Hb Lepore tienen presentación hematimérica y morfológica semejante a la b-Tal-het.
En el laboratorio hematológico, el diagnóstico de los síndromes b-Tal es más accesible que el de los a-Tal.
Anemia sideroblástica
Las anemias sideroblásticas constituyen un grupo heterogéneo de cuadros caracterizados por acúmulo de hemosiderina en las mitocondrias de los eritroblastos (sideroblastos en anillo). Las anemias sideroblásticas congénitas son raras. Entre ellas se encuentran la anemia sideroblástica ligada al cromosoma X (XLSA) y la variante asociada con ataxia (XLSA/A).
La XLSA resulta de la mutación de la δ-ácido aminolevulínico sintetasa 2 (ALAS2), y la XLSA/A es consecuencia de mutaciones en el gen del transportador mitocondrial ABC7 (23).
Las XLSA constituyen un grupo de desórdenes severos caracterizados por una inadecuada formación del Hemo. El consecuente aumento del Fe de depósito altera el estado de óxido reducción de la célula (24). En los casos graves, esta anemia es microcítica (VCM 50-60 fL) e hipocrómica y se acompaña de anisocitosis, poiquilocitosis y target cell.
Otros desórdenes
Hay dos grupos de anemias microcíticas raras, unas por defectos en el Hemo y las otras por defectos genéticos en el metabolismo del Fe.
Entre los defectos en el Hemo se encuentran, además de XLSA (y la variante asociada a ataxia), otras formas de anemia sideroblástica por mutaciones en genes GLRX5 y SLC25A38, hipotransferrinemia hereditaria, aceruloplasminemia hereditaria y porfirias (porfiria congénita eritropoyética y protoporfiria eritropoyética) (25).
Entre los defectos genéticos del metabolismo del Fe se encuentran: mutaciones en DMT1, anemia por deficiencia de Fe refractaria al Fe (IRIDA), y mutaciones en el gen STEAP3 (six transmembrane epithelialantigen of the prostate 3) el cual produce la ferrirreductasa endosomal correspondiente. La forma de herencia en las tres es autosómica recesiva. La primera es la que tiene más elevados niveles TfR, y la última es la que tiene mayor Ft (25).
Anemia ferropénica versus b talasemia menor
HEMATIMETRÍA
Para evaluar la hematimetría hay que considerar el uso de diferentes contadores hematológicos y su nivel tecnológico.
La OMS estableció valores límites de la Hb por debajo de los cuales se considera que una persona tiene anemia. Los valores límites en el hombre, mujer, embarazo y niños entre 5 a 11 años son de 13 g/dL, 12 g/dL, 11 g/dL, 11,5 g/dL, respectivamente. Por debajo de esos valores, a cada uno de estos grupos, se lo considera con anemia. En los niños menores a 5 años, los niveles de Hb varían de acuerdo con el mes y año de vida.
Los parámetros esenciales para la evaluación inicial de la anemia son el recuento de eritrocitos (GR), Hb y hematocrito (Hto). A partir de éstos se definen los índices hematimétricos de amplio uso como el VCM, HCM y concentración de la hemoglobina corpuscular media (CHCM). El VCM evalúa el tamaño del eritrocito. La HCM mide el contenido de Hb de cada eritrocito. La CHCM expresa la cantidad de Hb en todo el paquete globular. VCM y HCM permiten definir y clasificar la anemia como m-H. El CHCM tiene significancia en anemias con presencia de esferocitos, pero no es útil para el estudio de la m-H. En éstas, la CHCM empieza a descender cuando la anemia es muy marcada.
La ADH cursa con menor Hb que la b-Tal-het. En la Tabla III se presentan valores de Hb provenientes de diferentes grupos de trabajo. En todos los casos la diferencia fue significativa entre los controles normales y los dos grupos patológicos (ADH y b-Tal-het).
Tabla III. Hemoglobina.

En la b-Tal-het, el VCM generalmente es menor que en la ADH. En la b-Tal-het, los valores de la Hb y del VCM son constantes en cada persona, y pueden depender de la mutación. Los VCM y los HCM son ligeramente mayores en las b+-Tal-het que en las b0-Tal-het. Por ejemplo, en mujeres con b+-Tal-het la media del VCM fue de 64,19±1,25 fL vs 61,12±0,76 fL en mujeres con b0-Tal-het, y las medias de HCM fue de 19,66±0,32 pg vs 18,89±0,19 pg, respectivamente(26).
Los índices VCM y HCM en a-Tal-het también son significativamente diferentes con respecto a los normales. Nillakupt et al (27) consideran que, ante un VCM menor a 75 fL y un HCM menor a 25 pg habría que investigar síndromes a-Tal.
Por otro lado, Killip et al (28) postulan que pacientes con anemia y VCM menores a 74 fL tienen alta probabilidad de presentar ADH.
Por otro lado, en la ADH, tanto el nivel de Hb como el VCM no son constantes y varían con el grado de deficiencia de Fe (29) (30). Pacientes con valores “casi indetectables” de Ft, pueden tener valores de Hb inferiores a 7 g/dL y VCM por debajo de 60 fL. Adehossi et al (29) hallaron que mujeres en edad fértil con ADH severa tenían una Hb media de 6,9 g/dL (3,3-8,6 g/dL), y VCM medio de 78,3 fL (límite inferior 63 fL). La ADH, cuando es severa, puede tener VCM semejantes a los de los síndromes Tal menores. Esto debe considerarse al establecer un diagnóstico diferencial.
También el HCM es un buen parámetro para la detección de portadores de a-Tal-het y b-Tal-het (31).
Las a-Tal, con un gen comprometido, tienen VCM (media 81,2 fL) y HCM (media 26,2 pg) cercanos al límite inferior normal. Cuando están involucrados dos genes, el VCM (media 71,6 fL) y el HCM (media 22,9 pg) están disminuidos (32).
El ancho de distribución de eritrocitos o dispersión del tamaño celular o índice de Bessman (ADE o RDW) es una medida de la anisocitosis (de la heterogeneidad del tamaño de los eritrocitos). El ADE, junto al VCM, colabora en la clasificación de anemias y contribuye a realizar el diagnóstico diferencial (33).
Tanto en las a-Tal-het como en las b-Tal-het, el ADE es relativamente normal ya que todos los eritrocitos “responden” a la misma información genética. En cambio, en las Tal moderadas a severas, el ADE está aumentado como resultado de otros mecanismos fisiopatológicos que se ponen en juego. Entre ellos está el aumento importante de reticulocitos los cuales tienen un VCM mayor.
En la ADH, el ADE está incrementado (34). Esto implica que hay distintas poblaciones eritrocitarias, con diferentes niveles de hemoglobinización. A medida que avanza la deficiencia de Fe, va descendiendo el aporte del mismo para la síntesis de Hb, lo cual se refleja en el tamaño del eritrocito. O sea que coexisten eritrocitos de diferente edad y contenido de Hb, lo que produce incremento del ADE.
El ADE comienza a aumentar en la etapa latente, antes que desciendan la Hb y el VCM, por lo cual se lo considera un marcador medio en la evaluación de la deficiencia de Fe (35). Cuando la anemia está instaurada, el ADE sigue aumentado. O sea, que el ADE también se incrementa con la severidad de la anemia (36).
En la Tabla IV se presentan valores de VCM y ADE obtenidos por distintos grupos de trabajo.
Tabla IV. VCM y ADE.

Hace varias décadas se postularon diferentes índices de microcitosis para contribuir con el diagnóstico diferencial entre ADH y Tal-het. Estos índices son resultado de cálculos matemáticos a partir de los valores de diferentes parámetros: índice de Mentzer (MI) (VCM/ GR), función discriminatoria England Fraser (E&F) (VCM-GR-5 x H-3,4), Srivastava (Sr) (HCM/GR). Estos índices son mayores en la ADH que en las Tal-het.
Otros autores propusieron más fórmulas matemáticas para orientar al diagnóstico diferencial: índice de Shine y Lal (S&L) (VCM2 x HCM/100), índice de Green y King (G&K) (VCM2 x ADE/Hb x 100), índice de ADE (ADEI) (ADE x VCM/GR), y el índice Sirdah (VCM-GR-3 x Hb) (37-42).
Okan et al (43) compararon pacientes con b-Tal-het y ADH a través de diferentes fórmulas. Independientemente de la severidad de la ADH, estos autores consideran que el índice S&L, seguido de G&K, son los más eficientes para discriminar b-Tal-het y ADH.
Ferrara (44) considera que, para la diferenciación entre estas dos anemias, no hay un único índice y/o fórmula de elección. Encontró que la mayor sensibilidad correspondió al ADEI (78,9%), la más alta especifidad a E&F (99,1%) y la más alta eficiencia a G&K (80,2%). Demir (45) concluye que GR y ADEI fueron los mejores para la discriminación, pero para Ntaios (46) el mejor índice sería G&K.
Para AlFadhli (47), el E&F presentó altas especificidad y sensibilidad para discriminar ADH con Tal-het (a y b), y para Bruccoleri (48), el MI es el mejor.
En experiencia de esta autora (30), GR, el VCM (más que el HCM), y el MI son buenos orientadores iniciales en la discriminación de las dos patologías.
En la db-Tal-het, VCM, HCM y ADE son un poco más elevados que en b-Tal-het, y el ADE correlaciona con el nivel de Hb fetal (49).
Algunos contadores hematológicos de alta tecnología, brindan nuevos parámetros para evaluar los eritrocitos: índice de microcitosis e hipocromía (índice m-H) y porcentaje de células hipocrómicas.
El índice m-H (porcentaje células microcíticas – porcentaje células hipocrómicas) es un parámetro de alta sensibilidad y especificidad. Es una herramienta útil para el diagnóstico diferencial de anemias m-H. En pacientes con índice m-H mayor a 11,5, habría que pensar más en una Tal-het que en una ADH y, en consecuencia, en estos individuos sería aconsejable continuar el estudio para una Tal (37) (50).
El aumento del porcentaje de células hipocrómicas, a diferencia de otros marcadores, es un indicador directo de deficiencia funcional de Fe, o sea, de su disponibilidad para la eritropoyesis (51).
El recuento de reticulocitos es superior en la b-Tal-het que en la ADH, pero en ésta, los reticulocitos son más inmaduros, lo que sugiere cierto grado de eritropoyesis ineficiente (49). Cuando la ADH es severa, el recuento de reticulocitos puede aumentar.
Dentro de las b-Tal-het, las b0 tienen mayor recuento reticulocitario que las b+(26).
El recuento de reticulocitos no da información del estado de deficiencia de Fe, pero el contenido de Hb en los reticulocitos (HCMr) es indicador del grado de hemoglobinización (11), a semejanza del porcentaje de células hipocrómicas.
La HCMr es inferior en la ADH que en APC (52).
En la deficiencia de Fe, el descenso de HCMr antecede a la manifestación de la anemia y aumenta, aproximadamente, un 10% a las 48 horas de iniciado el tratamiento con Fe (antes del aumento del recuento de reticulocitos).
Ullrich et al (53) consideran que la medida del descenso de la HCMr es mejor que el nivel de Hb total para detectar un estado de deficiencia de Fe. EL aumento de células hipocrómicas y el descenso de HCMr son importantes para sospechar ADH (54). Valores inferiores a 26 pg de HCMr son sugestivos de deficiencia funcional de Fe (55) (56). HCMr correlaciona con la saturación de Tf y Ft.
En síntesis:
b-Tal-het vs ADH:
Mayores valores de GR, Hb, recuento de reticulocitos en b-Tal-het que en ADH
b-Tal-het vs ADH:
Menores valores de VCM, HCM, ADE, índices de microcitosis en b-Tal-het que en ADH
Cuando la ADH es muy severa pueden no cumplirse algunos de estos puntos. Los valores de algunos parámetros (VCM, recuento de reticulocitos, por ejemplo) son semejantes a los de la b-Tal-het.
En los portadores a-Tal, los valores son semejantes a los de los b-Tal-het.
El porcentaje de células hipocrómicas y HCMr son útiles para evaluar estados de deficiencia de Fe.
Metabolismo del hierro
El estudio de la ADH se realiza por medio de parámetros tradicionales que evalúan el metabolismo del Fe, como Fe en sangre, Tf (y su capacidad de saturación) y Ft.
En la ADH están disminuidos el Fe y la saturación de la Tf, y la Tf está aumentada.
Como el Fe plasmático tiene elevada variabilidad biológica entre individuos, pero también en el propio individuo (edad, dieta, momento del día, determinadas situaciones fisiológicas como embarazo, entre otras), hay que disponer de otros parámetros de laboratorio.
Estos son necesarios para evaluar diferentes situaciones donde está alterado el metabolismo del Fe como en la ADH (y sus etapas previas), en la APC, y en patologías que cursan con sobrecarga de Fe. Cuando la saturación de la Tf es menor a 10% podría indicar deficiencia franca de Fe (ADH). Pero si el porcentaje es entre 10 y 15%, no sólo podría indicar deficiencia de Feper se, sino también infección o carcinoma (57).
La Ft contiene aproximadamente el 13% del Fe presente en el organismo, lo que representa, en condiciones fisiológicas, más del 50% del Fe de reserva. Es un almacén de hierro “movilizable” y protege a las células del daño oxidativo del Fe libre.
Hasta la fecha, el descenso de la Ft sérica (evaluación del Fe de depósito) constituye el marcador más precoz con el que cuenta el laboratorio, para evaluar estadíos de deficiencia de Fe (desde el inicio de la etapa prelatente).
En las b-Tal hay una inefectiva eritropoyesis y una desregulación del metabolismo del Fe, lo que resulta en aumento del Fe de depósito (58). Aunque en la b-Tal-het, los niveles en sangre periférica de Fe, Tf y Ft no tienen diferencias significativas con respecto al normal, en el estado homocigota hay importantes aumentos de Fe plasmático y de depósito (ferritina y hemosiderina).
En algunos individuos, la ADH puede coexistir con procesos inflamatorios. En estas circunstancias, hay que recurrir a otros parámetros del metabolismo del Fe, ya que la determinación de la Ft no es suficiente para una evaluación correcta y completa.
El TfR es una glicoproteína de transmembrana que se encuentra principalmente en la superficie de los eritroblastos y controla la incorporación del Fe para la hemoglobinogénesis. Los receptores solubles de transferrina (sTfR) son producto del clivaje de los TfR que están presentes en los eritroblastos y reticulocitos (59).
La expresión de los sTfR está regulada por diferentes factores, y principalmente, por el nivel de Fe de depósito y la eritropoyesis (60).
En la deficiencia de Fe tisular se produce un incremento del número de TfR.
La concentración plasmática de Ft y el índice sTfR / log Ft son útiles como marcadores de eritropoyesis en anemias con descenso del Fe plasmático. Los sTfR son útiles tanto como marcadores relativamente precoces de deficiencia de Fe como en la diferenciación entre la ADH y la APC (34). Tanto el sTfR como el índice sTfR/ log Ft están más elevados en ADH y en APC con ferropenia, que en APC pura.
Los sTfR también están aumentados en los síndromes talasémicos. Aumentan en respuesta a la actividad eritropoyética, pero ese incremento no es tan importante en los portadores, donde la hemólisis es leve y compensada.
Jayaranee (61) halló los siguientes valores de sTfR: controles normales 1,53+0,8 mg/L, ADH 5,53+8,76 mg/L y Tat-het 1,64+1,02 mg/L. Algo semejante se observa (30) en la concentración de los sTfR (nmoles/L) en individuos normales, con ADH y con b-Tal-het: 18,4±2,2; 113,1±20,0; 41,6±5,5, respectivamente. En ambos estudios se encontró que la diferencia fue altamente significativa entre el grupo con ADH y los controles normales.
Los sTfR son marcadores medios de la deficiencia de Fe, ya que comienzan a aumentar cuando ha comenzado a disminuir el Fe de los depósitos.
El valor de los TfR depende del grado de deficiencia de Fe. Y siguen aumentando una vez que la anemia se manifiesta y agrava. Dentro del grupo con ADH (30), el paciente con menor Hb (5,6 g/dL) presentó el valor más alto de receptores (400 nmoles/L).
Se compararon valores de Ft y sTfR en embarazadas que presentaban ADH y embarazadas no anémicas. Los valores de Ft fueron menores (24,9±10,48 μg/L vs 31,03±9,98 μg/L, p=0,001), y los de los sTfR, más altos (1,40±0,0802 μg/mL vs 1,08±0,641 μg/mL, p=0,019), en las embarazadas con ADH con respecto a embarazadas no anémicas, respectivamente (62).
La determinación de los sTfR es importante no sólo porque colabora en la discriminación entre ADH y otras anemias microcíticas, sino también porque brinda una información adicional relacionada con la intensidad de la deficiencia de Fe.
En síntesis:
ADH vs b-Tal-het:
Menores valores de Fe, Ft, saturación Tf en ADH que en b-Tal-het
ADH vs b-Tal-het:
Mayores valores de Tf, sTfR en ADH que en b-Tal-het
Los sTfR son útiles para evaluar la intensidad de la deficiencia de Fe, incluso ante depleción o ausencia de los depósitos de Fe.
La evaluación del metabolismo del Fe permite el diagnóstico de ADH y su grado de severidad.
La determinación conjunta de Fe sérico, saturación Tf, Ft y sTfR permite hacer diagnóstico diferencial entre ADH y APC.
Estudio de la hemoglobina
La Hb del adulto está constituida por HbA (a2b2) (mayor de 97,0%), HbA2 (a2d2) (1,6 a 3,0%) y Hb fetal (a2g2) (hasta 1,0%).
Como el estudio de determinados parámetros del metabolismo del Fe es fundamental para el diagnóstico de ADH, el estudio de las fracciones de la Hb y su cuantificación, son imprescindibles para definir la b-Tal-het.
En la b-Tal-het, la HbA2 está incrementada, y la Hb fetal está normal o ligeramente aumentada.
En portadores de b-Tal, Origa et al (63) encontraron valores de HbA2 mayores a 3,5% y de Hb fetal entre 0,5 y 4,0%.
En un estudio anterior (30), los valores de HbA2 y de Hb fetal fueron de 4,5±0,1% y 1,6±0,1% en los pacientes con b-Tal-het, 1,9±0,1% y 1,2±0,1% en los pacientes con ADH, y 2,5±0,1% y 0,8±0,1% en los controles normales, respectivamente.
Bragos et al (26) analizaron las fracciones de Hb en pacientes con diferentes mutaciones de b-Tal-het. No encontraron diferencias en los porcentajes de HbA2 ni de Hb fetal, entre las b0 y las b+-Tal. La media de HbA2, en los hombres, para b0 fue de 5,38±0,15%, para b+ 5,40±0,19%, y, en las mujeres, de 5,21±0,09% y 5,24±0,15%, respectivamente. La Hb fetal en hombres con b0 fue 2,42±0,25, para b+Tal 1,75±0,32 y, en mujeres, de 2,51±0,15 y 2,04±0,26, respectivamente.
Dentro de los síndromes b-Tal hay variantes genéticas menos frecuentes, como la bd-Tal y la Hb Lepore. En ambas existe, además de descenso de síntesis de cadenas b, disminución de cadenas d. En el caso de Hb Lepore, también está presente esta Hb, la cual tiene anomalías estructurales, producto de un gen híbrido originado por el entrecruzamiento desigual de porciones de los genes d y b. Los heterocigotas de estas dos variantes tienen un fenotipo semejante a la b-Tal-het en cuanto a hematimetría y morfología. Por esto, para el diagnóstico diferencial entre síndromes b Tal heterocigotas, es fundamental la identificación y cuantificación de las fracciones de Hb. En las db-Tal-het la HbA2 está normal o ligeramente disminuida y la Hb fetal está aumentada. En Hb Lepore, la HbA2 está normal o ligeramente disminuida y la Hb fetal, normal o aumentada. La Hb Lepore representa un 5 a 15% de la Hb total y tiene movilidad intermedia entre la HbA y HbA2, en medio alcalino.
En el portador a-Tal, la HbA2 está normal o ligeramente disminuida.
El estudio molecular de las b-Tal es utilizado para confirmar y/o completar el diagnóstico dado por la electroforesis. Pero para el diagnóstico de la a-Tal menor es imprescindible la biología molecular ya que no hay cambios en los estudios electroforéticos (64).
En la ADH, la HbA2 está ligeramente disminuida como consecuencia del descenso de Fe para la síntesis de Hb. La Hb fetal está normal o ligeramente aumentada. El nivel de HbA2 disminuye con el descenso de la Hb total y del Fe sérico, y con el aumento de los sTfR. Estos cambios se relacionan con el descenso de la hemoglobinogénesis.
La Hb fetal no es decisiva en el discernimiento entre ADH y b-Tal-het. Pero siempre hay que considerar el porcentaje de esta Hb para la diferenciación entre síndromes b-Tal-het (b vs bd).
También hay otras causas de aumento de Hb fetal no relacionadas con las anemias m-H, como anemias hemolíticas, embarazo, determinadas medicaciones, leucemia mieloide crónica juvenil, algunos carcinomas, entre otras (65-69).
En síntesis:
Mayor valor de HbA2 en b-Tal-het que en ADH.
La Hb fetal no es un buen parámetro para diferenciar las dos patologías.
Tener en cuenta los síndromes a-Tal-het y otros síndromes b-Tal-het (db-Tal-het y Hb Lepore het) para realizar el diagnóstico diferencial. Se deben investigar las fracciones de Hb y, si es necesario, realizar el estudio molecular.
Estrés oxidativo
Las especies reactivas del oxígeno se generan permanentemente en las células (radical hidroxilo •OH, anión superóxido O2-, peróxido de hidrógeno H2O2, oxígeno singlete 1O2.). Varios elementos esenciales para la nutrición del ser humano (Fe, Cu, Mn, Co, Mo) existen en múltiples estados de oxidación y son capaces de catalizar reacciones del estrés oxidativo. Para minimizar esto, se expresan varias proteínas (Ft, Tf, lactoferrina, haptoglobina, ceruloplasmina) que aseguran que estos metales permanezcan escondidos (crípticos) (70). Hay, además, importantes sistemas antioxidantes: enzimáticos y no enzimáticos. Los no enzimáticos consisten en moléculas scavenger (basureros, barredoras, atrapadoras de luz) que son producidas endógenamente (glutatión, ubiquinoles, ácido úrico). Otras provienen de la dieta como carotenoides, vitaminas C, E, selenio, zinc, manganeso. Los sistemas enzimáticos están constituidos por superóxido dismutasa (SOD), catalasa, glutatión peroxidasa (GPX), glucosa-6-fosfato deshidrogenasa (G6PD) y glutatión reductasa (71).
En las anemias m-H, donde está disminuido el contenido de Hb dentro del eritrocito, el daño oxidativo juega un rol importante en la patogénesis de las mismas. La Hb celular, target de los procesos oxidativos, está disminuida. Por lo tanto, otras macromoléculas, como los ácidos grasos insaturados de la membrana, son sustratos sensibles a la peroxidación (72-76).
El estrés oxidativo está en relación inversa con el valor de la Hb. Las ADH y Tal sufren daño oxidativo (descenso de la defensa antioxidante y aumento de marcadores de estrés oxidativo) el cual trae como consecuencia menor sobrevida celular,
En la Tal mayor, el daño oxidativo (77) es mucho más severo que en el heterocigota ya que, además de la hipocromía, hay aumento significativo de cadenas a globinas libres, del Hemo y del Fe (78).
En la literatura se encuentran diferentes estudios que analizan el estrés oxidativo en anemias m-H y hay controversias referidas tanto a las actividades enzimáticas encontradas como al grado de peroxidación lipídica(79) (80).
Estas divergencias entre los estudios se dan especialmente en ADH. Podrían ser consecuencia de la heterogeneidad en los grupos de pacientes seleccionados, con distintos grados de deficiencia de Fe. Estos grados de deficiencia están en relación con la efectividad del ensamble del Fe con las porfirinas y/o con la cantidad de Fe disponible y necesario para la síntesis de hemoproteínas como la catalasa (81). Altun et al (82) evaluaron dos grupos de individuos con deficiencia de Fe, con y sin anemia. En los pacientes sin anemia, los niveles de lipoperoxidación y de las actividades enzimáticas fueron más cercanos al grupo control normal.
Tanto los pacientes con ADH como los que tienen b-Tal-het presentan aumento significativo de la lipoperoxidación (30) (74-76) (79). La lipoperoxidación, en eritrocitos, está aumentada un 78% en el grupo con ADH y en un 20% en el grupo con b-Tal-het, con respecto a los normales (30). Otros autores hallaron que la lipoperoxidación fue mayor en la b-Tal-het que en la ADH y que en db heterocigota (83), y sin diferencia significativa con pacientes con HbE heterocigota (76).
Se obtuvieron actividades de G6PD significativamente incrementadas (p<0,001) en ADH (14,3±1,0 U/gHb) y b-Tal-het (14,0±0,5 U/gHb) con respecto al normal (8,2+0,3 U/gHb). En cambio, la capacidad antioxidante total del plasma (media+DE) estuvo significativamente disminuida (p<0,001) en ADH (142±7 µM) y b-Tal-het (140±11 µM) con respecto al normal (207±8 µM). No hubo diferencia entre ADH y b-Tal-het ni para G6PD y ni para la capacidad antioxidante total del plasma (30).
Diferentes autores discuten la respuesta a determinados tratamientos basados en la reducción del estado oxidativo de estas anemias m-H.
En general, ninguno de los parámetros de estrés oxidativo es el elegido para diferenciar entre estas ADH y b-Tal-het. Pero lo importante es considerar el daño oxidativo que existe en ellas, compararlo con controles normales, e implementar algún tratamiento antioxidante(como con determinadas vitaminas) que atenúe dicho daño. Con esto se puede mejorar la estabilidad celular y el nivel de la Hb (84).
En síntesis:
Debido a las diferencias encontradas en la bibliografía, dentro de los parámetros evaluadores de daño oxidativo no hay ninguno que sea ideal para diferenciar entre las dos anemias. Pero es importante analizarlos, en cada una, para evaluar la severidad de la anemia y la mejor respuesta al tratamiento.
Morfología eritrocitaria
A partir de las diferentes alteraciones morfológicas observadas en el frotis de sangre periférica, se puede inferir el grado de severidad de la anemia y qué parámetros de laboratorio pueden estar más alterados.
Los acantocitos y target cells son consecuencia de cambios de lípidos de la membrana. La presencia de punteado basófilo, importante en los pacientes talasémicos, está relacionada con el descenso de la actividad de la 5´pirimidin nucleotidasa. Los dacriocitos se observan en trastornos de la eritropoyesis, en disfunciones medulares, entre otros desórdenes (85) (86). Los esquistocitos, células mordidas y esferocitos pueden indicar estrés oxidativo y hemólisis (83) (87-89).
Las microcitosis e hipocromía están presentes en todos los pacientes con ADH y en todos los pacientes con b-Tal-het.
En el 100% de los pacientes con b-Tal-het, las microcitosis e hipocromía siempre están acompañadas por otras características morfológicas. Los acantocitos, target cells, punteado basófilo están presentes en un número importante de pacientes, y se observan esquistocitos, dacriocitos, excentrocitos, esferocitos, en menor porcentaje de ellos.
A diferencia de los pacientes talasémicos, no todos los pacientes con ADH tienen cambios en la morfología eritrocitaria, aparte de las microcitosis e hipocromía. De acuerdo con el grado de deficiencia de Fe (heterogeneidad de la población), los pacientes con ADH presentan determinadas alteraciones morfológicas y en diferente porcentaje. En ADH, la ausencia de cambios morfológicos eritrocitarios, en general se relaciona con mayores niveles de Hb, VCM y HbA2 y recuento normal de reticulocitos. Los pacientes con niveles más bajos de HbA2, relacionados con menor hemoglobinogénesis, presentan eliptocitosis, acantocitosis, punteado basófilo, target cells y esquistocitos (90).
Las diferentes alteraciones morfológicas están presentes, en mayor porcentaje, en portadores b-Tal que en pacientes con ADH: acantocitos (70,2 vs 53,2%), punteado basófilo (72,3 vs 17,0%), target cells (46,8 vs 19,1%), esquistocitos (36,2 vs 14,9%), dacriocitos (17,0 vs 12,8%), esferocitos (8,5 vs 4,3%), excentrocitos (6,3 vs 4,3%), respectivamente (30).
Cuando la anemia es muy severa, con daño oxidativo de importancia, en ambas patologías aparecen células mordidas.
En los pacientes con b-Tal-het las alteraciones morfológicas responden a las diferentes mutaciones. Los pacientes heterocigotas con la mutación CD39 (b0) tienen mayor porcentaje de acantocitos y punteado basófilo que aquéllos con IVS-I-nt110 (b+).
En la a-Tal-het los cambios morfológicos celulares son menores y menos frecuentes que en b-Tal-het.
En síntesis:
Mayores alteraciones morfológicas (tanto en tipo como en frecuencia) en b-Tal-het que en ADH:
En la ADH los cambios morfológicos aparecen y se acentúan con la severidad de la anemia (al ir disminuyendo la Hb); en las b-Tal-het dependen de la mutación.
Discusión y Conclusiones
Dentro de la clasificación morfológica, las anemias microcíticas, generalmente hipocrómicas (VCM menor a 80 fL; HCM menor a 27 pg), son las más frecuentes en la práctica clínica. En ellas está afectada la hemoglobinogénesis (alteración en el ensamble del Hemo con las globinas). Son anemias microcíticas e hipocrómicas las anemias por deficiencia de Fe, las Tal y algunas anemias sideroblásticas. Las APC generalmente son normocíticas pero, en determinadas circunstancias, pueden ser microcíticas. Es importante establecer el diagnóstico, buscar parámetros de laboratorio que permitan una detección precoz de la enfermedad, y establecer una adecuada y sencilla discriminación entre ellas.
Se han analizado diferentes aspectos de la ADH y de la b-Tal-het, por ser las más prevalentes en nuestro medio. Los marcadores fundamentales de la ADH son la ferremia y la Ft, y los de la b-Tal-het, el aumento de HbA2.
Existen otros parámetros que se pueden utilizar para diferenciar estas dos patologías. Es importante analizar los datos aportados por el contador hematológico, los cuales pueden contribuir a orientar al estudio a una u otra anemia. Esto también evita hacer estudios innecesarios y/o demora en llegar al diagnóstico y tratamiento correspondiente. La observación del frotis de sangre periférica es un complemento necesario. De acuerdo con la complejidad del laboratorio o a las características del paciente (edad, embarazo, infecciones, entre otras) se pueden realizar otros estudios que permitan diagnosticar, evaluar la severidad y diferenciar estas dos entidades.
Se exponen las principales relaciones entre ambas patologías:
ADH: <ferremia, Ft, saturación de la Tf que en b-Tal-het
b-Tal-het: >recuentos de GR y reticulocitos, Hb, HbA2, alteraciones morfológicas que en ADH
ADH: >ADE, índices de microcitosis, Tf, sTfR que en b-Tal-het
b-Tal-het: <VCM, HCM que en ADH
En la Tabla V se expone cómo varían determinados parámetros del laboratorio de ambas patologías con respecto a los valores de referencia.
Tabla V. Parámetros de laboratorio.

Como conclusión final, se propone un algoritmo de las anemias m-H más frecuentes a partir del valor del Fe sérico. Se incluye ADH (y no los estados previos) y síndromes Tal heterocigotas, pero no los homocigotas (Figura 2).
Después de excluir ADH, APC y síndromes b-Tal-het, hay que investigar síndromes a talasémicos por biología molecular. Estos son la causa de la mayoría de los casos de microcitosis inexplicadas (91).
1. Baltierra D, Harper T, Jones MP, NauDC. Hematologic Disorders: Anemia. FS Essent 2015 Jun; 433: 11-5. [ Links ]
2. Irwin JJ, Kirchner JT. Anemia in children. Am Fam Physician 2001 Oct 15; 64 (8): 1379-86. [ Links ]
3. Janus J, Moerschel SK. Evaluation of anemia in children. Am Fam Physician 2010 Jun 15; 81 (12); 1462-71. [ Links ]
4. Iolascon A, De Falco L, Beaumont C. Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme synthesis. Haematologica 2009 Mar; 94: 395–408.
5. Doty RT, Phelps SR, Shadle C, Sanchez-Bonilla M, Keel SB, Abkowitz JL. Coordinate expression of heme and globin is essential for effective erythropoiesis. Clin Invest 2015 Dec; 125 (12): 4681-91. [ Links ]
6. Montoya Romero J de J, Castelazo Morales E, Valerio Castro E, Velázquez Cornejo G, Nava Muñoz DA, Escárcega Preciado JA et al. Review by expert group in the diagnosis and treatment of anemia in pregnant women. Federación Mexicana de Colegios de Obstetricia y Ginecología. Ginecol Obstet Mex 2012 Sep; 80 (9): 563-80. [ Links ]
7. Durán M. Anemia por deficiencia de hierro: estrategias disponibles y controversias por resolver. Arch Argent Pediatr 2007; 105 (6): 488-90 [ Links ]
8. Encuesta Nacional de Nutrición y Salud (ENNyS). Anemia. La desnutrición oculta. [Internet]. Ministerio de Salud - Presidencia de la Nación 2008. Disponible en: http://www.datos.dinami.gov.ar/produccion/nutricion/materia/A1d.df. (Fecha de acceso 15 de mayo de 2016. [ Links ]
9. Lee CT, Jeng CJ, Yeh LS, Yen MS, Chen SM, Lee CL, et al. A double-blind, randomized, and active-controlled phase III study of Herbiron drink in the treatment of iron-deficiency anemia in premenopausal females in Taiwan. Food Nutr Res 2016 Jun 23; 60: 31047. [ Links ]
10. Zerga M. Anemia de los trastornos crónicos. Hematología 2004; 8 (2): 45-55. [ Links ]
11. Thomas DW, Hinchliffe RF, Briggs C, Macdoogall IC, Littlewood T, Cavill I. British Committee for Standards in Haematology: Guideline for the laboratory diagnosis of functional iron deficiency. Br J Haematol 2013; 161 (5): 639-48. [ Links ]
12. Ganz T. The role of hepcidin in iron sequestration during infections and in the pathogenesis of anemia of chronic disease. Isr Med Assoc J 2002; 4 (11): 1043-5. [ Links ]
13. Means RT Jr. Hepcidin and iron regulation in health and disease. Am J Med Sci 2013 Jan; 345 (1): 57-60. [ Links ]
14. Lou DQ, Nicolas G, Lesbordes C, Viatte L, Grimber G, Szajnert MF et al. Funcional differences between hepcidin 1 and 2 in transgenic mice. Blood 2004; 103 (7): 2816-21. [ Links ]
15. De Domenico I, Ward DM, Kaplan J. Hepcidin regulation: ironing out the details. J Clin Invest 2007 Jul; 117 (7): 1755-8. [ Links ]
16. Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood 2005 Oct 15; 106(8): 2884-9. [ Links ]
17. Giorgi G. Estudio de la movilización intracelular del hierro y su desregulación. Universidad Nacional del Sur; 2016. Tesis Doctoral. [ Links ]
18. Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest 2005 Aug; 115 (8): 2079-82. [ Links ]
19. De las Heras Florez S, Pérez Hernández LM. Hemoglobinopatías diagnosticadas en el área sanitaria del Hospital Universitario Nuestra Señora de Candelaria de Santa Cruz de Tenerife durante un año. An Med Interna (Madrid) 2008 Feb 25; (2): 61-6. [ Links ]
20. Ruano-Ravina A, Jato-Díaz M, Cerdá-Mota T. Cribado neonatal de hemoglobinopatías. Una reflexión sobre su aplicación en España. Med Clin (Barc) 2006; 126 (9): 337-40. [ Links ]
21. Huang SW, Xu Y, Liu XM, Zhou M, Lii GF, An BQ, et al. The Prevalence and Spectrum of α-thalassemia in Guizhou Province of South China. Hemoglobin 2015; 39 (4): 260-3.
22. Ruiz Delgado G, Vázquez Garza E, Ibarra B, Perea FJ, Gómez Almaguer D. Hemoglobinopatía H: comunicación de un caso identificado en Monterrey, Nuevo León. Med Int Mex 2008; 24 (1): 76-8. [ Links ]
23. Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007 Aug 15; 110 (4): 1353-8. [ Links ]
24. Astner I, Schulze JO, van den Heuvel J, Jahn D, Schubert WD, Heinz DW. Crystal structure of 5-aminolevulinate synthase, the first enzyme of hemebiosynthesis, and its link to XLSA in humans. EMBO J 2005 Sep 21; 24 (18): 3166-77. [ Links ]
25. Bruno M, De Falco L, Iolascon A. How I diagnose non-thalassemic microcytic anemias. Seminars Hematol 2015 Oct; 52 (4): 270-8. [ Links ]
26. Bragós IM, Noguera NI, Raviola MP, Milani Á. Genética molecular de beta talasémicos heterocigotas: Interrelación con parámetros hematológicos. Rev Cubana Hematol Inmunol Hemoter [Internet] 2005 Abr; 21(1): Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-02892005000100005&lng=. (Fecha de acceso 10 de julio de 2016). [ Links ]
27. Nillakupt K, Nathalang O, Arnutti P, Jindadamrongwech S, Boonsiri T, Panichkul S et al. Prevalence and hematological parameters of thalassemiain Tha Kradarn subdistrict Chachoengsao Province, Thailand. J Med AssocThai 2012 May; 95 Suppl 5: S 124-32. [ Links ]
28. Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician 2007 Mar 1; 75 (5): 671-8. [ Links ]
29. Adehossi E, Malam-Abdou B, Andia A, Djibrilla A, Sani Beydou S, Brah S et al. Geophagy associated with severe anemia in non-pregnant women: A case series of 12 patients. Rev Med Interne 2016 Mar 23. pii: S0248-8663(16)00087-4. [Epub ahead of print]. [ Links ]
30. Aixalá M. Anemia microcítica: relación entre perfil lipoproteico sérico y estrés oxidativo. Universidad de Buenos Aires 2008. Tesis doctoral. [ Links ]
31. Pranpanus S, Sirichotiyakul S, Srisupundit K, Tongsong T. Sensitivity andspecificity of mean corpuscular hemoglobin (MCH): for screening alpha-thalassemia-1 trait and beta-thalassemia trait. J Med Assoc Thai 2009 Jun; 92 (6): 739-43. [ Links ]
32. Origa R, Moi P, Galanello R, Cao A. Alpha-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016 2005 Nov 1[updated 2013 Nov 21]. [ Links ]
33. Bessman JD, Feinstein DI. Quantitative anisocytosis as a discriminant between iron deficiency and thalassemia minor. Blood 1979 Feb; 53 (2): 288-93. [ Links ]
34. Fernández García N, Aguirrezabalaga González B. Anemias en la infancia. Anemia ferropénica. Bol Pediatr 2006; 46: 311-7. [ Links ]
35. Bessman JD, Gilmer PR Jr, Gardner FH. Improved classification of anemias by MCV and RDW. Am J Clin Pathol 1983 Sep; 80 (3): 322-6. [ Links ]
36. Aulakh R, Sohi I, Singh T, Kakkar N. Red cell distribution width (RDW) in the diagnosis of iron deficiency with microcytic hypochromic anemia. Indian J Pediatr 2009 Mar; 76 (3): 265-8. [ Links ]
37. Urrechaga E, Hoffmann JJ, Izquierdo S, Escanero JF. Differential diagnosis of microcytic anemia: the role of microcytic and hypochromic erythrocytes. Int J Lab Hematol 2015 Jun; 37 (3): 334-40. [ Links ]
38. Sirdah M, Tarazi I, Al Najjar E, Al Haddad R. Evaluation of the diagnostic reliability of different RBC indices and formulas in the differentiation of the beta-thalassaemia minor from iron deficiency in Palestinian population. Int J Lab Hematol 2008 Aug; 30 (4): 324-30. [ Links ]
39. Beyan C, Kaptan K, Ifran A.Predictivevalue of discrimination indices in differential diagnosis of iron deficiency anemia and beta-thalassemia trait. Eur J Haematol 2007 Dec; 79 (6): 554. [ Links ]
40. Nalbantoglu B, Güzel S, Büyükyalçın V, Donma MM, Güzel EÇ, Nalbantoglu A, et al. Indices used in differentiation of thalassemia trait from iron deficiency anemia in pediatric population: are they reliable? Pediatr Hematol Oncol 2012 Aug; 29 (5): 472-8. [ Links ]
41. Sargolzaie N, Miri-Moghaddam E. A local equation for differential diagnosis of β-thalassemia trait and iron deficiency anemia by logistic regression analysis in Southeast Iran. Hemoglobin 2014 Aug; 26: 1-4.
42. Pornprasert S, Panya A, Punyamung M, Yanola J, Kongpan C. Red cell indices and formulas used in differentiation of β-thalassemia trait from iron deficiency in Thai school children. Hemoglobin 2014; 38 (4): 258-61.
43. Okan V, Cigiloglu A, Cifci S, Yilmaz M, Pehlivan M. Red cell indices and functions differentiating patients with the beta-thalassaemia trait from those with iron deficiency anaemia. J Int Med Res 2009 Jan-Feb; 37 (1): 25-30. [ Links ]
44. Ferrara M, Capozzi L, Russo R, Bertocco F, Ferrara D. Reliability of red blood cell indices and formulas to discriminate between beta thalassemia trait and iron deficiency in children. Hematology 2010 Apr; 15 (2): 112-5. [ Links ]
45. Demir A, Yarali N, Fisgin T, Duru K, Kara A. Most reliable índices in differentiation between thalassemia trait and iron deficiency anemia. Pediatr Int 2002 Dec; 44 (6): 612-6. [ Links ]
46. Ntaios G, Chatzinikolaou A, Saouli Z, Girtovitis F, Tsapanidou M, Kaiafa G et al. Discrimination indices as screening tests for beta-thalassemic trait. Ann Hematol 2008 Jan; 87 (1): 61-2. [ Links ]
47. AlFadhli SM, Al-Awadhi AM, AlKhaldi D. Validity assessment of nine discriminant functions used for the differentiation between iron deficiency anemia and thalassemia minor. J Trop Pediatr 2007 Apr; 53 (2): 93-7. [ Links ]
48. Bruccoleri F, Zepponi E, Balucanti F, Ciciarelli U, Di Donna G,Di Folco S, et al. Determination of the diagnostic value of erythrocyte discrimination índices (Mentzer and Srivastava) and of glicerol lysis time (GLT 50) in microcytosis. Quad Sclavo Diagn 1982 Mar; 18 (1): 67-75. [ Links ]
49. Velasco-Rodríguez D, Alonso-Domínguez JM, González- Fernández FA, Villarrubia J, Sopeña M, Abalo L et al. Reticulocyte parameters of delta beta thalassaemia trait, beta thalassaemia trait and iron deficiency anaemia. J Clin Pathol 2016 Feb; 69 (2); 149-54. [ Links ]
50. Urrechaga E, Borque L, Escanero JF. The role of automated measurement of red cell subpopulations on the Sysmex XE 5000 analyzer in the differential diagnosis of microcytic anemia. Int J Lab Hematol 2011 Feb; 33 (1): 30-6. [ Links ]
51. Thomas C, Thomas L. Biochemical markers and hematologic indices in the diagnosis of functional iron deficiency. Clin Chem 2002 Jul; 48 (7): 981-2. [ Links ]
52. Schaefer DM, Stokol T. The utility of reticulocyte indices in distinguishing iron deficiency anemia from anemia of inflammatory disease, portosystemic shunting, and breed-associated microcytosis in dogs. Vet Clin Pathol 2015 Mar; 44 (1): 109-19. [ Links ]
53. Ullrich C, Wu A, Armsby C, Rieber S, Wingerter S, Brugnara C, et al. Screening healthy infants for iron deficiency using reticulocytehemoglobin content. JAMA 2005 Aug 24; 294 (8): 924-30. [ Links ]
54. David O, Grillo A, Ceoloni B, Cavallo F, Podda G, Biancotti PP, et al. Analysis of red cell parameters on the Sysmex XE 2100 and ADVIA 120 in iron deficiency and uraemic chronic disease. Scand J Clin Lab Invest 2006; 66 (2): 113-20. [ Links ]
55. Mittman N, Sreedhara R, Mushnick R, Chattopadhyay J, Zelmanovic D, Vaseghi M, et al. Reticulocyte hemoglobin content predicts functional iron deficiency in hemodialysis patients receiving rHuEPO. Am J Kidney Dis 1997 Dec; 30 (6): 912-22. [ Links ]
56. Brugara C, Schiller B, Moran J. Reticulocyte hemoglobin equivalent (Ret He) and assessment of iron-deficient states. Clin Lab Haematol 2006 Oct; 28 (5): 303-8. [ Links ]
57. Cailliat MC, Fink NE. Algoritmos de laboratorio para el estudio del estado del hierro. Acta Bioquím Clín Latinoam 2013; 47 (3): 507-22. [ Links ]
58. Ginzburg Y, Rivella S. b-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011 Oct 20; 118 (16): 4321-30. [ Links ]
59. Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin Chim Acta 2003 Mar; 329 (1-2): 9-22. [ Links ]
60. R´zik S, Loo M, Beguin Y. Reticulocyte transferrin receptor (TfR) expression and contribution to soluble TfR levels. Haematologica 2001 Mar; 86 (3): 244-51. [ Links ]
61. Jayaranee S, Sthaneshwar P. Serum soluble transferrin receptor in hypochromic microcytic anaemia. Singapore Med J 2006 Feb; 47 (2): 138-42. [ Links ]
62. Sharma JB, Bumma SD, Saxena R, Kumar S, Roy KK, Singh N et al. Cross sectional, comparative study of serum erythropoietin, transferrin receptor, ferritin levels and other hematological índicesin normal pregnancies and iron deficiency anemia during pregnancy. Eur J Obstet Gynecol Reprod Biol 2016 May 20; 203: 99-103. [ Links ]
63. Origa R. Beta-Thalassemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. Gene Reviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.2000 Sep 28 [updated 2015 May 14]. [ Links ]
64. Brancaleoni V, Di Pierro E, Motta I, Cappellini MD. Laboratory diagnosis of thalassemia. Int J Lab Hematol 2016 May 16. [Epub ahead of print]. [ Links ]
65. Anderson UD, Gram M, Ranstam J, Thilaganathan B, Kerström B, Hansson SR. Fetal hemoglobin, a1-microglobulin and hemopexinare potential predictive first trimester biomarkers for preeclampsia. Pregnancy Hypertens 2016 Apr; 6 (2): 103-9. [ Links ]
66. Wolk M, Martin JE. Fetal haemopoiesis marking low-gradeurinary bladder cancer. Br J Cancer 2012 Jul 24; 107 (3): 477-81. [ Links ]
67. Kar R, Saxena R, Pati HP. Hereditary spherocytosis with high fetal hemoglobin: an interesting case. Hemoglobin 2008; 32 (5): 520-3. [ Links ]
68. Unal S, Chui DH, Gumruk F. Fanconi´s anemia effect or sickle cell anemia effect. That is the question. Hemoglobin 2015; 39 (4): 287-9. [ Links ]
69. Almeida CB, Souza LE, Leonardo FC, Costa FT, Werneck CC, Covas DT et al. Acute hemolytic vascular inflammatory processes are prevented by nitric oxide replacement or a single dose ofhydroxyurea. Blood 2015 Aug 6; 126 (6): 711-20. [ Links ]
70. Chan A, Chow C, Chiu D. Interaction of antioxidants and their implication in genetic anemia. Proc Soc Exp Biol Med 1999. 222 (3): 274-82. [ Links ]
71. Rodríguez Perón JM, Menéndez López JR, Trujillo López Y. Radicales libres en la biomedicina y estrés oxidativo. Rev Cubana Med Milit 2001; 30 (1): 36-44. [ Links ]
72. Brandão MM, Castro Mde L, Fontes A, Cesar CL, Costa FF, Saad ST. Impaired red cell deformability in iron deficient subjects. Clin Hemorheol Microcirc 2009; 43 (3): 217- 21. [ Links ]
73. Jain SK, Yip R, Hoesch RM, Pramanik AK, Dallman PR, Shohet SB. Evidence of peroxidative damage to the erythrocyte membrane in iron deficiency. Am J Clin Nutr 1983 Jan; 37 (1): 26-30. [ Links ]
74. Lazarte SS, Mónaco ME, Jimenez CL, Ledesma Achem ME, Terán MM, Issé BA. Erythrocyte catalase activity in more frequent microcytic hypochromic anemia: Beta-Thalassemia trait and iron deficiency anemia. Adv Hematol 2015; 2015: 343571. [ Links ]
75. Madhikarmi NL, Murthy KR. Antioxidant enzymes and oxidative stress in the erythrocytes of iron deficiency anemic patients supplemented with vitamin. Iran Biomed J 2014; 18 (2): 82-7. [ Links ]
76. Manisha D, Banerjee D, Talukdar G, Bhattacharya DK. A study of spectrin and lipid peroxidation of red blood cell membrane in thalassaemia carrier. Indian J Clin Biochem 1999 Jul; 14 (2): 207-12. [ Links ]
77. Claster S, Wood JC, Noetzli L, Carson SM, Hofstra TC, Khanna R et al. Nutritional deficiencies in iron overloaded patients with hemoglobinopathies. Am J Hematol 2009 Jun; 84 (6): 344-8. [ Links ]
78. Koskenkorva-Frank T, Weiss G, Koppenol W, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radical Biology and Medicine 2013; 65: 1174-94. [ Links ]
79. Coghetto Baccin A, Lauerman Lazzaretti L, Duarte Martins Brandao V, Manfredini V, Peralba MC, Silveira Benfato M.Oxidative stress in older patients with iron deficiency anaemia. J Nutr Health Aging 2009 Aug; 13 (8): 666-70. [ Links ]
80. Isler M, Delibas N, Guclu M, Gultekin F, Sutcu R, Bahceci M et al. Superoxide dismutase and glutathione peroxidase in erythrocytes of patients with iron deficiency anemia: effects of different treatment modalities. Croat Med J 2002 Feb; 43 (1): 16-9. [ Links ]
81. Ozcicek F, Aktas M, Türkmen K, Coban TA, Cankaya M The investigation of plasma glucose-6-phosphate dehydrogenase, 6-phoshogluconate dehydrogenase, glutathione reductase in premenauposal patients with iron deficiency anemia. Pak J Med Sci 2014 Jul; 30 (4): 809-913. [ Links ]
82. Altun D, Kurekci AE, Gursel O, Hacıhamdioglu DO, Kurt I, Aydın A et al. Malondialdehyde, antioxidant enzymes, and renal tubular functions in children with iron deficiency or iron-deficiency anemia. Biol Trace Elem Res 2014 Oct; 161 (1): 48-56. [ Links ]
83. Vives Corrons JL, Miguel-García A, Pujades MA, Miguel-Sosa A, Cambiazzo S, Linares M et al. Increased susceptibility of microcytic red blood cells to in vitro oxidative stress. Eur J Haematol 1995 Nov; 55 (5): 327-31. [ Links ]
84. Madhikarmi NL, Murthy KR. Antioxidant enzymes and oxidative stress in the erythrocytes of iron deficiency anemic patients supplemented with vitamins. Iran Biomed J 2014: 18 (2): 82-7. [ Links ]
85. Grignaschi V, Díaz N, Alonso M, Lardo M, Lucero G. Citomorfología y citoquímica hemáticas. 2º Ed. Buenos Aires: Ediciones Britania; 1983. [ Links ]
86. Vives Corrons JL, Pujades MA, Aguilar I, Bascompte J, Jou J, Rozman C et al. Pyrimidene 5´nucleotidase and several other red cell enzyme activities in beta-thalassaemia trait. Br J Haematol 1984, 56 (3): 483-94. [ Links ]
87. David O, Vota MG, Piga A, Ramenghi U, Bosia A, Pescarmona GP. Pyrimidine 5'-nucleotidase acquired deficiency in beta-thalassemia: involvement of enzyme-SH groups in the inactivation process. Acta Haematol 1989; 82 (2): 69-74. [ Links ]
88. Clark MR, Aminoff MJ, Chiu DT, Kuypers FA, Friend DS. Red cell deformability and lipid composition in two forms of acanthocytosis: enrichment of acanthocytic populations by density gradient centrifugation. J Lab Clin Med 1989 Apr; 113 (4): 469-81. [ Links ]
89. Yokoyama A, Nakamaki T, Yamada K, Koike M, Tomoyasu S, Hirayama N et al. Beta 0-thalassemia trait (IVS-I-1 G-- >T) in a Japanese family. Intern Med 1993 Nov; 32 (11): 865-8. [ Links ]
90. Rodgers MS, Chang CC, Kass L. Elliptocytes and tailed poikilocytes correlate with severity of iron-deficiency anemia. Am J Clin Pathol 1999 May; 111 (5): 672-5. [ Links ]
91. Loonat SB, Naran NH, Thein SL, Alli NA. Alpha-thalassaemia trait as a cause of unexplained microcytosis in a South African population. S Afr Med J 2016 Feb 22; 106 (3): 276-9. [ Links ]
Recibido: 18 de julio de 2016
Aceptado: 20 de julio de 2017

