










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa bioquímica clínica latinoamericana
versión impresa ISSN 0325-2957versión On-line ISSN 1851-6114
Acta bioquím. clín. latinoam. vol.52 no.2 La Plata jun. 2018
BIOQUÍMICA CLÍNICA
Cupremia y colesterolemia como factores de riesgo en pacientes con enfermedad de Alzheimer*
Plasma levels of copper and cholesterol as risk factors in Alzheimer disease patients
Cupremia e colesterolemia como fatores de risco em pacientes com doença de Alzheimer
Carina Grisela Jomñuk1a, Diana Olga Cristalli2b, Nathalie Arnal3c, Carlos Alberto Marra4d, Leandro Vecchio Dezillio5e, Natalia Lausada6f
1 Médica Especialista en Neurología (UNLP).
2 Médica Especialista Jerarquizado y Consultor en Neurología (UNLP).
3 Dra. en Ciencias de la Salud (UNLP).
4 Dr. en Bioquímica Clínica (UNLP).
5 Médico Veterinario (UNLP).
6 Dra. en Medicina Veterinaria (UNLP).
a Hospital San Roque de Gonnet (La Plata).
b Profesora Adjunta con semi-dedicación de la Cátedra de Neurología de la Fac. Cs. Médicas de la UNLP, Investigadora Categoría IV Integrante del Programa de Incentivos para Docentes-Investigadores (Ministerio de Educación de la Nación, UNLP).
c Jefe de Trabajos con dedicación exclusiva a la investigación de la Cátedra de Bioquímica y Biología Molecular, Fac. Cs. Médicas de la UNLP. Investigadora Integrante del Programa de Incentivos para Docentes-Investigadores (Ministerio de Educación de la Nación, UNLP), Investigadora Asistente del CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas) en el INIBIOLP (Instituto de Investigaciones Bioquímicas de La Plata).
d Investigador Principal del CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas), Profesor con dedicación exclusiva de la Cátedra de Bioquímica y Biología Molecular (Fac. Cs. Médicas de la UNLP), Investigador Categoría I del Programa de Incentivos para Docentes-Investigadores (Ministerio de Educación de la Nación, UNLP), Investigador del INIBIOLP (Instituto de Investigaciones Bioquímicas de La Plata, Fac. Cs. Médicas de la UNLP)
e Becario Doctoral en el LTO (Laboratorio de Trasplante de Órganos), Fac. de Cs. Médicas de la UNLP.
f Directora de Proyecto Acreditado para el Programa de Incentivos a Docentes-Investigadores del Ministerio de Educación de la Nación. Profesora Adjunta con dedicación exclusiva e Investigadora del LTO (Laboratorio de Trasplante de Órganos) de la Fac. de Cs. Médicas de la UNLP.
* INIBIOLP (Instituto de Investigaciones Bioquímicas de La Plata), CCT - La Plata, CONICET-UNLP, Cátedra de Bioquímica y Biología Molecular, Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Calles 60 y 120 (1900) La Plata, Argentina.
CORRESPONDENCIA Prof. Dr. CARLOS ALBERTO MARRA INIBIOLP (Instituto de Investigaciones Bioquímicas de La Plata), CCT - La Plata, CONICET-UNLP, Cátedra de Bioquímica y Biología Molecular, Facultad de Ciencias Médicas, Universidad Nacional de La Plata, Calle 60 y 120 (1900) LA PLATA, Argentina. E-mail: contactocarlos@hotmail.com
Resumen
Se estudiaron 241 personas, 119 controles y 122 pacientes con enfermedad de Alzheimer (EA) subagrupados en tres categorías de acuerdo con el estadio clínico de la dolencia, con el objetivo de investigar la influencia de niveles elevados de cobre libre y colesterol plasmático como factores de riesgo para la EA. Las conclusiones obtenidas de los resultados indicaron que los pacientes expuestos a una combinación de alto colesterol y de cobre no unido a ceruloplasmina tuvieron mayor proporción de marcadores de estrés oxidativo (carbonilos proteicos, sustancias reactivas al tiobarbiturato, glutatión oxidado y descenso de antioxidantes totales en sangre), conjuntamente con un incremento de HDL-colesterol peroxidado y lipoproteína "a" que correlacionó con la gravedad de su cuadro. Lo mismo sucedió con la relación entre péptidos amiloides (1-40) y (1-42) en plasma y los valores del mini-test de estado cognitivo (MMSE). Se halló que una función de adición de efectos que cuantificó el daño por cobre libre y colesterol oxidado resultó directamente proporcional a la pérdida de desempeño cognitivo estimada por medio del MMSE. Esta función es de fácil determinación y puede considerarse un nuevo biomarcador para estudiar riesgo en poblaciones expuestas, apoyar el diagnóstico clínico, o evaluar la eficacia de estrategias terapéuticas en pacientes con EA.
Palabras clave: Colesterolemia; Cupremia; Enfermedad de Alzheimer; Desempeño cognitivo; Estrés oxidativo; Proteínas amiloides; Ceruloplasmina; Sustancias reactivas al ácido tiobarbitúrico; Colesterol HDL oxidado.
Abstract
Alzheimer disease (AD) patients (122) compared to control subjects (119) were studied to determine the role of chronic exposure of hypercholesterolemic plasma levels and free copper (not bound to ceruloplasmin) as biomarkers of progression for AD. Oxidative stress parameters, lipid profile, amyloid levels, and cognitive status were studied in all participants. Conclusions obtained indicated that patients exposed simultaneously to free copper and increased cholesterol levels present higher indicators of oxidative stress (protein carbonyls, thiobarbituric acid-reactive substances, decreased total antioxidant activity in plasma and elevated oxidized HDL-cholesterol). Lipoprotein "a" concentrations also correlated with the clinical progression of the disease. The ratio amyloid ß(1-40)/ß(1-42) in plasma inversely correlated with the cognitive performance estimated by the Mini-Mental State Examination Test (MMSE). A defined function that weighs the contribution of the deleterious effect produced by combined free copper and Ox-HDL-cholesterol exposure directly correlated with the loss of cognitive performance. Thus, this biomarker could be a new tool for the screening of large populations under risk, or may be a useful way to estimate the efficacy of therapeuti approaches in patients suffering AD.
Keywords: Plasma cholesterol; Plasma copper; Alzheimer disease; Cognitive performance; Oxidative stress; Amyloid proteins; Ceruloplasmin; Thiobarbituric acid-reactive substances; Oxidized-HDL cholesterol.
Resumo
Foram estudadas 241 pessoas, 119 controles e 122 pacientes com doença de Alzheimer (DA), agrupados em três categorias de acordo com o estágio clínico da doença, com o objetivo de investigar a influência de níveis elevados de cobre livre e colesterol plasmático como fatores de risco para a DA. As conclusões obtidas a partir dos resultados indicaram que os pacientes expostos a uma combinação de colesterol alto e de cobre não ligados à ceruloplasmina apresentaram maior proporção de marcadores de estresse oxidativo (carbonilos proteicos, substâncias reativas ao tiobarbiturato, glutationa oxidada e diminuição dos antioxidantes totais no sangue ), juntamente com um aumento da HDL-colesterol peroxidado e lipoproteína "a" que correlacionou com a gravidade de sua condição. O mesmo aconteceu com a relação entre os peptídeos amilóides (1-40) e (1-42) em plasma e os valores do mini-teste do estado cognitivo (MMSE). Verificou-se que uma função de adição de efeitos que quantificou o dano por cobre livre e colesterol oxidado resultou diretamente proporcional à perda de desempenho cognitivo estimada através do MMSE. Esta função é fácil de determinar e pode ser considerada um novo biomarcador para estudar o risco em populações expostas, apoiar o diagnóstico clínico ou avaliar a eficácia de estratégias terapêuticas em pacientes com DA.
Palavras-chave: Colesterolemia; Cupremia; Doença de Alzheimer; Desempenho cognitivo; Estresse oxidativo; Proteínas amilóides; Ceruloplasmina; Substâncias reativas ao ácido tiobarbitúrico; Colesterol HDL oxidado.
Abreviaturas
bA1-40: amiloide beta fragmento 1-40
bA1-42: amiloide beta fragmento 1-42
ANOVA: análisis de la varianza
APP: péptido precursor de beta amiloide
CPs: carbonilos proteicos
CRP: ceruloplasmina
CT: colesterol total
Cu: cobre total
DCL: deterioro cognitivo leve
EA: enfermedad de Alzheimer
FAE: función de adición de efectos
FRAP: capacidad antioxidante total
HDL-C: colesterol de lipoproteínas de alta densidad
LDL-C: colesterol de lipoproteínas de baja densidad
Lp(a): lipoproteína "a"
MMSE: Mini Mental State Examination
MTs: metalotioneínas
NCBC: cobre no ligado a ceruloplasmina
Ox-LDL: lipoproteína de baja densidad peroxidada
PBS: buffer fosfato sódico 50 mM; pH: 7,40
PCRC: porcentaje de cambio respecto al control
TBARS: sustancias reactivas al tiobarbiturato
X: promedio de la muestra
(Otras abreviaturas utilizadas esporádicamente se aclaran en el mismo texto, o en las figuras y tablas).
Introducción
Muchos avances socioeconómicos y científico-tecnológicos han provocado un tremendo impacto en la prolongación de la expectativa de vida durante la última centuria. Sin embargo y paradójicamente, el hombre ha desarrollado un estilo de vida más sedentario, asociado a malos hábitos dietarios y a la exposición de multitud de contaminantes medioambientales que él mismo genera de manera irresponsable y descontrolada. Estos factores de presión epigenéticos han inducido un incremento desproporcionado de complicaciones derivadas de enfermedades metabólicas y neurodegenerativas. Lo que en los últimos años se denominaba "enfermedad neurológica degenerativa", actualmente y según sea la dolencia referida, se convirtió en un fenómeno cada vez más precisamente conocido de alteración en el clivaje de proteínas, o de la tasa de síntesis y configuración de las mismas, defectos en el almacenaje o distribución de diferentes sustancias neurotransmisoras, mutaciones genéticas específicas causadas por alteraciones tóxicas, nutricionales, o debidas a infección por virus lentos o por un agente transmisible no viral (como los priones).
Está ampliamente aceptado que el desempeño cognitivo predice con gran exactitud la chance de institucionalización y sobrevida (1) por lo que se han redoblado los esfuerzos para estudiar este tipo de desórdenes. Por otro lado, se asiste a una verdadera revolución en el tratamiento de las mismas dado que cada vez más neurólogos, o profesionales de ciencias afines, hablan de un tratamiento farmacológico y uno no farmacológico, refiriéndose a intervenciones para mejorar la calidad de vida de los pacientes (que hace 20 años eran considerados prácticamente no tratables justamente por sufrir enfermedades neurodegenerativas). Se trabaja intensamente en todo el mundo para elucidar los factores medioambientales que determinan el progreso de estas enfermedades, su prevención, y el desarrollo de estrategias paliativas y/o curativas. La enfermedad de Alzheimer (EA) es un trastorno neurológico primario, progresivo, degenerativo, e incurable que afecta fundamental-mente al cerebro y que lleva a una creciente pérdida de múltiples funciones, lo cual finalmente determina la falta total de identidad de la propia persona. Fue descripta por primera vez en 1907 por el Dr. Alois Alzheimer y hoy constituye la forma de demencia más frecuente en los países occidentales con un 2% de incidencia y una prevalencia de entre el 56 y el 70% (dependiendo de los autores) siendo la causa más frecuente de demencia irreversible. Se cree que un 11% de las personas mayores de 60 años padecen alguno de los signos centinelas de la EA (2). En Argentina se observa una prevalencia importante de enfermos de EA calculados en algo más de 500.000 (3-5). Este dato per se indica la gravedad de este problema médico. Existen varios factores de riesgo para la EA y se la considera una enfermedad de etiología multifactorial. Como entidad clínica es muy heterogénea y puede estar íntimamente vinculada a factores genéticos debidos a mutaciones en los cromosomas 1, 14, 19 y 21. La interacción del genoma del paciente con el entorno resulta crucial en su establecimiento y progresión (presión epigenética). No existe una clasificación aceptada para la EA, pero dada su heterogeneidad, se pueden identificar varios subtipos. La gran mayoría de los casos son esporádicos, pero un 10 a 15% son claramente genéticos con herencia autosómica dominante. La edad es el factor que más influye en la heterogeneidad clínica ya que la EA de comienzo precoz se asocia a deterioro más rápido, mayor frecuencia de formas familiares, y mayor pérdida neuronal en núcleos subcorticales (3-5). Conforme se sobrepasan los 60 años la prevalencia de la enfermedad se duplica cada 5 años llegando a ser mayor al 56% entre las personas que superan los 85 años (6). Los antecedentes familiares de parientes de primer grado con EA incrementan 4 veces el riesgo de padecerla. Lamentablemente se carece en la actualidad de una prueba de laboratorio que tenga la suficiente especificidad y sensibilidad como para realizar el diagnóstico diferencial de la EA de manera temprana y confiable. No obstante, algunos estudios permiten evaluar la progresión de la enfermedad o la eficacia de los tratamientos. Por lo antes dicho parece pertinente extender y profundizar las observaciones referentes tanto al mecanismo íntimo por el cual el daño neurológico acontece, como a su diagnóstico y pronóstico, de manera de disponer de estrategias para mitigarlo y contar con elementos de juicio confiables que puedan (potencialmente) ser empleadas en la práctica clínica. En este sentido se ha dirigido la atención hacia factores de riesgo novedosos o emergentes en busca de una mayor información que esclarezca el acuciante conflicto del incremento observado en su incidencia y al mismo tiempo resulten ser útiles en la prevención de la enfermedad (y/o en la evaluación de su terapia). Hoy día se sabe que la etiopatogenia de la EA involucra a metales pesados de transición como el Cu y a su sistema homeostático, como así también al colesterol y sus catabolitos o productos de peroxidación. A medida que el cerebro envejece ocurren progresivos eventos pro-oxidativos e inflamatorios que son característicos de la pérdida de neuronas/sinaptosomas y de glía (7-9). Es bien conocido que el cerebro es el tejido más sensible al insulto pro-oxidativo. Desde hace muchos años quedó demostrada la presencia de productos de oxidación de biomoléculas tales como los carbonilos proteicos (oxidación de proteínas) y peróxidos lipídicos co-localizando con placas seniles y neuríticas (10-13). Por otra parte, el Cu se destaca por ser un metal de transición con una muy importante actividad redox (14). Se lo encuentra en trazas en un sinnúmero de biomoléculas y enzimas críticas para el normal desarrollo de la fisiología del cerebro, pero al mismo tiempo, representa una
importante fuente endógena de radicales libres (15- 18). Dentro del sistema nervioso central (SNC) el Cu unido a ceruloplasmina (CRP) es minoritario respecto del libre (19-21), situación completamente opuesta a lo que acontece con el cobre circulante en sangre periférica (22). Por lo tanto, es razonable hipotetizar que el exceso de Cu no unido a CRP (NCBC) –o la disrupción del equilibrio entre Cu libre y NCBC– es un factor crítico en la etiopatogénesis de los daños cerebrales de base pro-oxidativa (20-32). Ahora es bien conocido que los iones Cu se hallan íntimamente involucrados en la etiopatogénesis de las enfermedades neurodegenerativas humanas, en especial en el mal de Parkinson (PD), en las demencias a cuerpos de Lewy, y explícitamente en el mal de Alzheimer (20)(32)(34-37). El Cu cataliza reacciones pro-oxidativas de tipo Fenton (38) que generan especies altamente reactivas atacando a biomoléculas claves en el metabolismo o en la estructura de biomembranas (ADN, proteínas y especialmente lípidos) (38) (39) y está asociado tanto a la gliosis reaccional o activación microglial como al estrés oxidativo en el SNC que desencadena señales de muerte neuronal programada redox-dependientes (24)(29)(39-42). En relación a esta cuestión se destacan los hallazgos del grupo Italiano de Rosana Squitti dedicado específicamente al estudio de los efectos del Cu en la etiología de la EA (43-48). Experimentalmente se probó que pequeñas cantidades de Cu inducen la acumulación de péptidos bA y la formación de placas seniles en cerebro de conejos hipercolesterolémicos pero no ocurre lo mismo en conejos normocolesterolémicos (50-52). Como es sabido, los péptidos amiloides bA se generan por proteólisis de su precursor APP (una proteína trans-membrana) a través de las enzimas b- y γ- secretasas (53)(54). Se ha demostrado que ambas enzimas se encuentran asociadas a rafts (o "balsas" de lípidos de membrana) muy enriquecidas en colesterol y que probablemente sea este lípido el responsable de aumentar la actividad de ambas enzimas (55-59). Aparentemente el exceso de colesterol induce la formación de placas amiloides o "seniles" constituidas principalmente por agregación de péptidos bA. El grupo de Zambón (60) demostró la estrecha correlación entre el deterioro cognitivo y la hipercolesterolemia cuando se trata de pacientes que la padecen por largos períodos (o muy tempranamente en su vida) o cuando existe alguna disfunción en los receptores de LDL colesterol. Así, el deterioro cognitivo presenta una incidencia del 21,3% en pacientes con hipercolesterolemia familiar vs. el 2,9% observado en una población testigo que no padeció este desorden (60). Por otro lado, también se sabe que el bA induce daño pro-oxidativo per se (60-62), y que se halla asociado a la progresión y al comienzo del daño neurodegenerativo (20)(65) (66). Numerosas investigaciones previas sugieren que existe una clara asociación entre el efecto pro-apoptótico de pequeñas cantidades de Cu y el nivel circulante de colesterol cuando éste último se halla por encima de los valores normales (10)(21)(55)(59)(60)(67-71). También es conocido que los complejos (bA)/Cu+2 de las placas amiloides pueden acumular colesterol y producir peróxido de hidrógeno y formas de colesterol oxidado (peroxi-colesteroles) los cuales son altamente neurotóxicos (72-76). Muchos investigadores consideran que el péptido precursor amiloide (APP) y los bA forman un sistema "homeostático" del exceso de Cu en el SNC ya que unen al metal con muy alta afinidad (34) (44)(78)(79).
La evidencia disponible hasta ahora parece sugerir que se requiere de la co-existencia de ambos factores (hipercolesterolemia y sobrecarga de cobre libre o NCBC) para que el daño neurológico efectivamente se produzca, y al parecer, este daño estaría determinado por la muerte celular programada mediante señales pro-oxidativas o redox-dependientes (69)(73)(74)(80- 85). A pesar de lo mencionado, no se ha esclarecido en humanos la relación precisa entre pequeñas cantidades de Cu, el exceso de colesterol, y el establecimiento de estrés oxidativo en el proceso de neurodegeneración que corresponde al curso clínico de la EA. Tampoco se sabe si la barrera hematoencefálica resulta dañada por estos factores de riesgo y si esto jugaría un papel importante en el proceso neurodegenerativo. En este punto, resulta importante aclarar que la sobrecarga de Cu es un hecho mucho más frecuente de lo sospechado, y que existen poblaciones expuestas a cantidades excesivas de este metal durante largos periodos de su vida en forma sub-signológica y sub-sintomática (4)(40) (83)(86-91). Por ejemplo, el Cu contenido en el agua de bebida de muchas regiones del mundo es causa suficiente de preocupación (20)(92-97). En cuanto al metabolismo lipídico en la EA, se sabe que el colesterol transportado por LDL se halla continuamente expuesto a un ambiente pro-oxidativo con abundante formación de especies radicalarias derivadas del oxígeno y nitrógeno principalmente (ROS y RNS), situación que en el sistema nervioso central se halla agravada por un extenuado sistema de defensa antioxidante enzimático y no enzimático. La peroxidación de los lípidos de estas partículas provoca la acumulación de aldehídos y otros productos de la fractura de la cadena hidrocarbonada de los ácidos grasos (malondialdehído y varias otras sustancias reactivas al tiobarbiturato o TBARS). A su vez, se oxidan y carbonilan los restos lisina de las cadenas laterales de los aminoácidos (con formación de los llamados carbonilos proteicos (PCs) (98-107). Esta transformación es responsable de una respuesta inflamatoria local en el endotelio y de la activación de macrófagos (94)(108-112). En la superficie de las LDL oxidadas (OX-LDL) aparecen nuevos epítopes que finalmente provocan una respuesta autoinmune sobre todo en pacientes crónicos o mal controlados (94)(113).
En suma, la EA estaría íntimamente vinculada a un cuadro de estrés oxidativo caracterizado por dishomeostasis del cobre y del metabolismo del colesterol. Por esa razón, el objetivo de este estudio fue determinar varios marcadores bioquímicos asociados a estos tres aspectos para luego analizar si existe entre ellos una vinculación que pueda darles valor clínico como elementos pronósticos o evaluativos de las estrategias terapéuticas a que se sometan los pacientes con EA. La hipótesis operacional enuncia entonces que los contenidos de cobre libre (NCBC) y de colesterol en sangre periférica humana presentan correlación positiva con el marcador de amiloidogénesis bA y negativa con el de desempeño cognitvo evaluado por medio del MMSE (Mini-Mental State Examination test).
Materiales y Métodos
Químicos y drogas de referencia. Las drogas utilizadas para hacer todas las determinaciones se adquirieron a Sigma Chem. Co. (EE.UU. o Buenos Aires), Merck Co. (Darmstadt, Alemania), Fluka (Berlín, Alemania), Anedra (Buenos Aires, Argentina) o Carlo Erba (Milán, Italia). El cobre empleado en forma de sulfato cúprico pentahidratado de calidad ultrapura (electrolítica grado máximo) lo suministró Sigma-Aldrich. Todos los estándares utilizados en la calibración de las técnicas se adquirieron a los fabricantes de los equipos o se compraron en grado ultrapuro a las fuentes previamente mencionadas. Los gases (calidad 5 bandas) los proveyó la empresa AGA de La Plata, Argentina.
Grupos experimentales. Se trabajó con dos grupos de personas, uno control designado con la letra "C", y un grupo de pacientes con diagnóstico de enfermedad de Alzheimer ("EA"), estos últimos divididos en tres subgrupos denominados leves o "L-EA" con MMSE promedio (X): 20,0±2,3; intermedios o "I-EA" (X: 14,5±1,7) y avanzados/severos o "S-EA" (X: 7,1±2,0). El X del MMSE del grupo control fue de 29,5±0,9. Los materiales utilizados en las determinaciones y los estudios efectuados se detallarán en los siguientes ítems.
Selección de controles y pacientes. Las poblaciones de pacientes y controles se seleccionaron durante los años 2013-2015 a partir de los consultorios externos de instituciones públicas y privadas del Gran La Plata siendo la mayoría de ellos asistidos en el Servicio de Neurología del Hospital San Roque de Gonnet. Los controles fueron ingresados a partir de consultorios externos de tres Hospitales Universitarios (San Martín, Gutiérrez y Rossi, de la UNLP) y de instituciones privadas, todos ellos derivados de controles rutinarios de salud. Se siguió un criterio de inclusión/exclusión en el cual se contempló la mayor cantidad de variables confusionales (114). En el caso de los controles, se hizo énfasis en descartar –mediante cuidadosa anamnesis y examen clínico y/o estudios complementarios previos al cuestionario de exclusión definitivo– a cualquier persona que tuviera un desorden genético o adquirido del metabolismo del cobre, un desorden nutricional de cualquier origen o siguiera una dieta no convencional (vegetarianos, ovo-lacto vegetarianos, veganos ambientalistas o sintagmáticos, etc.) o tuvieran una enfermedad neurológica o psiquiátrica de cualquier tipo. Fueron criterios de inclusión tener más de 50 años de edad, residir en la zona del Gran La Plata, y no padecer de ninguna enfermedad demostrable al examen clínico o a los estudios complementarios que se mencionarán en los criterios de exclusión. Para ser integrante del grupo de pacientes se debieron reunir los requisitos de edad y residencia antes mencionados y además padecer enfermedad de Alzheimer en alguno de sus estadios (inicial, intermedio o avanzado/severo) demostrado por los medios internacionalmente aceptados para el diagnóstico de esta patología, tener una evolución mínima de tres años desde el diagnóstico y encontrarse bajo un tratamiento farmacológico estable de al menos un año de duración previo al estudio y que se hallase comprendido dentro de los protocolos pautados o reconocidos por el consenso internacional para esta patología. Todos los integrantes fueron examinados para determinar su función renal y hepática, y su condición hematológica. Cuando las circunstancias lo justificaron se realizaron estudios complementarios de imágenes (RMN y TAC), doppler color en vasos carotídeos, endocraneales y vertebrales, y asimismo fueron examinados exhaustivamente desde el punto de vista neurológico. Seguidamente pasaron una evaluación por medio del test MMSE (115), que se ha demostrado que es la prueba más sencilla para llevar a cabo y al mismo tiempo más correlacionable con las baterías de evaluación neurocognitivas reconocidas internacionalmente (116-118) tales como Clinical Dementia Rating Score; Hachinski Score (119), Visuo-espatial Phonologic and Verbal Fluency Tests/AVLT: Auditory Verbal Learning Test, Clock Test, Addenbrook Test, y otras (120). Para agrupar a los pacientes en sub-categorías se los clasificó siguiendo las recomendaciones surgidas de la Global Deterioration Scale (121) y los criterios establecidos por el National Institute of Neurological and Communicative Disorders and Stroke (122), la Alzheimer´s Disease Association and Related Disorders (123) y también las establecidas por la American Psychiatric Association and Related Disorders (124). Para ambos grandes grupos (controles y pacientes) se convino seguir estrictamente las indicaciones dietarias e higiénico-sanitarias de modo de no alterar los hábitos y procederes que eran característicos en la vida de cada integrante, y se estableció que se debía tener un ayuno de 12 horas previo a la extracción de las muestras de sangre y a los exámenes clínicos y neurológicos. A todos se les practicó una encuesta alimentaria retrospectiva. Se consideraron criterios de exclusión enfermedades degenerativas osteoarticulares, patologías auto-inmunes de cualquier tipo (en especial lupus eritematoso o colagenopatías de origen diverso), neoplasias o displasias en curso (se admitieron solo aquellos casos en los cuales se había superado una enfermedad sin recidiva en los últimos 10 años), infecciones agudas o crónicas fúngicas, virales o bacterianas en curso (sífilis, HIV, dengue, fiebre amarilla, hepatitis virales de cualquier tipo, tuberculosis, micosis sistémicas de cualquier etiología tales como criptococosis, aspergillosis, candidiasis/candidemia, etc.), infección herpética diseminada, glomerulonefritis recidivantes, infestaciones de cualquier etiología estén o no en curso (Chagas, leishmaniasis, paludismo, toxoplasmosis, filariasis, histoplasmosis), asma y pacientes hiperérgicos sintomatológicos, enfisemáticos, pacientes con mucoviscidiosis, pacientes con dislipemias de origen congénito manifestadas durante la infancia o adolescencia, insuficientes hepáticos y/o renales, desnutridos, receptores de trasplantes de cualquier tejido u órgano sin importar el tipo de implante, cardiópatas congénitos, pacientes en el período post-quirúrgico de cualquier intervención, pacientes con endocrinopatías de cualquier etiología sin tratamiento, deportistas de élite o profesionales de cualquier rama del deporte, pacientes que recibieron tratamiento fibrinolítico por cualquier causa, mujeres con enfermedad poliquística de mama u ovario, pacientes convalecientes de trauma físico de cualquier tipo, pacientes edematosos o febriles en curso clínico, personas clínicamente sanas que consumieran preparaciones magistrales de cualquier tipo no recetadas tales como tisanas, formulaciones vitamínicas, oligoelementos, suplementos nutritivos/antioxidantes o preparados magistrales a base de ácidos grasos puros o en mezclas, esteroides de tipo anabólico o anti-inflamatorios esteroides o no esteroides administrados en forma aleatoria o no prescripta y alcohólicos o adictos/usuarios de cualquier droga adictiva incluso las denominadas menores (Cannabis, por ejemplo). Además de cuidar las indicaciones higiénico-sanitarias (alimentación, actividad física) se llevó un exhaustivo registro de la historia clínica de cada integrante en donde se consignó toda la medicación complementaria recibida y las intercurrencias o concomitancias que pudieran haber surgido a los fines de la validación estadística de los datos a correlacionar. Una vez practicada la preselección de grupos, el estudio excluyó a 89 candidatos y finalmente se realizó sobre un total de 241 personas (119 controles y 122 pacientes; a su vez, a partir del grupo EA se generaron tres sub-grupos integrados por 37 pacientes leves (L), 41 intermedios (I) y 44 severos (S)). Antes de ingresar al grupo que les correspondió todos firmaron directamente o por intermedio de su cuidador/representante un consentimiento informado tras la explicación de los objetivos, alcances, riesgos, y procederes del estudio de acuerdo con los contenidos de la ley Nac. Nº 25.326. La deserción voluntaria y/o exclusión provocada de las personas inicialmente ingresadas fue nula durante la realización del estudio.
Toma de muestras. Luego de los exámenes clínico-semiológico exhaustivos y complementarios, y del interrogatorio que contempló la recolección de datos demográficos y variables categóricas, se realizó la encuesta nutricional retrospectiva (adaptada de la versión oficial que fuera diseñada por el Ministerio de Salud; Encuesta Nacional de Nutrición y Salud (ENNyS), "Documento y Resultados"; Buenos Aires, 2007; URL: http://www.msal.gov.ar/htm/site/ennys/pdf/ENNyS_Documento-de-resultados-2007-II.pdf]). Los integrantes se sometieron a una extracción de sangre venosa mediante material descartable estéril y apirogénico tras 12 horas de ayuno. La sangre (aproximadamente 15 mL) se colocó en un tubo plástico de policarbonato de alta densidad, graduado con tapa a rosca de bioseguridad (Falcon Co. CA, EE.UU.) que contenía conservantes apropiados en concentraciones finales: buffer 50 mM fosfato sódico pH: 7,40 (PBS), heparina 2 UI/mL, EDTA disódico 5 mM, y butil-hidroxitolueno o BHT 10 mM, según correspondiera a cada tipo de determinación. Durante la colección y el transporte al laboratorio las muestras se mantuvieron a 4 oC y se las rotuló con un código para preservar el anonimato del paciente o del voluntario control. Luego de centrifugación por 20 min a 1.500 g se obtuvo el plasma. Una parte del mismo se trató con N-etilmaleimida a los fines que serán descriptos más adelante y el resto se congeló (a -80 oC) en un ultrafreezer bajo argón o nitrógeno de 5 bandas hasta que fue analizado. Además de las muestras de sangre cada integrante proporcionó una muestra del agua de bebida utilizada en su hogar para preparar alimentos y para beber. Esta muestra se recolectó en contenedores de policarbonato previamente lavados con solución de neocuproína (1 mM, Sigma Chem. Co., EE.UU.) a los fines de eliminar cualquier rastro de cobre que pudieran contener, se enjuagaron exhaustivamente con agua bidestilada y se secaron en estufa a 40 oC. Las muestras se tomaron haciendo correr el flujo de agua por 5 minutos (muestra "purgada", para descartar cobre añadido por la tubería u otros dispositivos del sistema en el agua estancada) siguiendo recomendaciones estándar (125). Asimismo, se analizaron varias muestras de agua dentro de la zona del Gran La Plata –zona de residencia de los integrantes del estudio– para determinar el contenido de cobre inorgánico proveniente del agua de red potable y de otras fuentes utilizadas en su reemplazo (agua de perforaciones particulares que alcanzaron al acuífero Epipuelche). Estas muestras se recolectaron de idéntica manera que las solicitadas a los integrantes del estudio y totalizaron 252 tomas.
Parámetros de química clínica y riesgo vascular calculado. Se determinaron varios compuestos presentes en sangre periférica para explorar las funciones hepática, renal, el estado hematológico, y el lipidograma básico, empleándose equipos comerciales de la firma Wiener Lab (Rosario, Argentina). Las muestras se procesaron siguiendo las instrucciones del fabricante. El riesgo vascular se calculó usando el software de acceso público del laboratorio Pfizer, URL: [https://www.pfizer.es/salud/servicios/calculadoras/calculadora_riesgo_cardiovascular.html]. Este resultado se lo ponderó con el estimador que se encuentra en el URL: [https://www.hipocampo.org/riesgovascular.asp] ajustándolo para un periodo de 10 años de vida.
Cupremia. Se empleó un método de referencia descripto por Berkovitch et al (126). A las muestras de plasma (100 μL) se les adicionó una mezcla formada con 4 mL de HNO3 y 1 mL de HClO4 (Carlo Erba, Italia; 99% pureza). Luego, se calentaron a 120 oC por 60 min en un bloque de calentamiento regulable. Los digestos se enfriaron y diluyeron con agua ultrapura (18 mW-cm; Carlo Erba). Esta solución fue ultrafiltrada usando una membrana Millipore de 0,22 μm (Milli-Q Purification System, de Millipore, CA, EE.UU.). Se inyectaron alícuotas de esta dilución en un espectrofotómetro de absorción atómica Perkin Elmer-1100B equipado con una lámpara catódica Perkin-Elmer (Perkin Elmer Corp., Norwalk, CT, EE.UU.). El ancho espectral fue de 1 nm. Todas las medidas se hicieron en tres canales simultáneos y en el modo de máximo espectral (324,1 nm). Las calibraciones se efectuaron usando una solución estándar de Cu(NO3)2 en HNO3 0,5 N (Tritrisol de Merck Co., Darmstadt, Alemania). Los coeficientes de variación intra- [(DE/X).100] e inter- [(DDE/X).100] experimentales fueron de 10,0 y 5,5%, respectivamente. La concentración de Cu en un pool de muestras de 55 voluntarios sanos de ambos sexos (promedio de edad: 56,0±5,5 años) fue de 802±55 μg/L (12,6 μM). Este resultado concordó con otros publicados previamente (126)(127). Las muestras de agua se analizaron previa ultrafiltración por membrana Millipore (0,22 μm) diluyéndolas luego con agua ultrapura (Carlo Erba) (1:20) y aspirándolas directamente en el equipo de absorción atómica (el bajo contenido en materia orgánica no hizo necesaria su mineralización previa y así se evitó agregar cobre en trazas que pudieran estar presente en los reactivos).
Determinación de metalotioneínas (MTs). Esta superfamilia de proteínas se determinó de acuerdo con la metodología de Bienengraber et al. (128). La técnica se basó en la capacidad de las MTs de captar Ag+ en una proporción estequiométrica conocida. La concentración de Ag+ pudo ser cuantificada utilizando un espectrofotómetro de absorción atómica. Por lo tanto, conociendo la estequiometría de formación del complejo se pudo inferir la concentración de MTs. Se tomaron alícuotas de muestra (60 μL de plasma) y se colocaron en tubos de borosilicato convenientemente rotulados agregándoles buffer glicina 0,5 M (pH 8,5) hasta completar 800 μL. Luego se agitaron y a continuación se añadieron 500 μL de solución de trabajo (20 μg Ag+/ mL). Se mezcló por inversión y se dejó incubar a temperatura ambiente por 5 min. Se continuó agregando 100 μL de un hemolisado de glóbulos rojos (aprox. 15 g de hemoglobina/dL) y se incubó en baño de agua a 100 oC por 1 ½ min. Los tubos se centrifugaron a 1.500 g por 5 min a temperatura ambiente. Se repitió la etapa anterior en el sobrenadante centrifugando de la misma manera. Se aspiró con cuidado el sobrenadante con una pipeta Pasteur y el aspirado se colocó en un tubo de vidrio con tapa a rosca. Luego se lo acidificó con HNO3 al 3% (500 μL cada 1,5 mL de muestra) antes de guardarlo a -17 oC y oscuridad hasta su uso. La concentración de Ag+ se midió por absorción atómica de la misma manera que se realizó para el Cu y utilizando el mismo equipo pero con el filtro específico para plata. Para realizar los cálculos se consideró que 1 μg de Ag+ equivale a 3,55 μg de MTs (128).
Determinación de ceruloplasmina (CRP). El método empleado (129) se basó en la actividad enzimática de la CRP que puede catalizar la conversión de p-fenilendiamina en un cromóforo púrpura cuya intensidad se pudo cuantificar en un espectrofotómetro a 550 nm y resultó ser directamente proporcional a la concentración de CRP. Las muestras (50 μL) se incubaron a 37 oC en un buffer acético/acetato de sodio 50 mM (pH 5,5) y las densidades ópticas se midieron en la fase inicial de la reacción (3 minutos) usando un equipo SpectraMax M2/M2e Multi-Detector Microplate Reader (Molecular Devices Analytical Technologies-Sunnyvale, CA, EE.UU.). Los coeficientes de variación intra- e inter-ensayo fueron de 8,0 y 4,2%, respectivamente. La velocidad inicial de reacción para cada muestra se calculó en forma automática y se convirtió en concentración de CRP por comparación con el estándar (Sigma Chem. Co., Buenos Aires, Argentina) el cual contenía 50 unidades de CRP-oxidasa/mg de proteína. Las mediciones de CRP en muestras de plasma de 55 personas sanas de ambos sexos de un promedio de edad de 56,0±5,5 años) arrojaron un X de 259±41 mg/L. Esto concordó con resultados previos obtenidos en humanos (130).
Cobre libre o no ligado a CRP (NCBC). Con los datos de Cu y CRP se calculó el porcentaje de Cu que no se halló unido a la CRP utilizando la siguiente fórmula debida a Twoney et al. (131): (([Cu] – 47,2 x [CRP]) x 100 / [Cu]). En este cálculo la concentración de Cu estuvo expresada en μmol/L y la de CRP en g/L. Con los datos del porcentaje de NCBC y el de cobre total se calcularon los resultados en ppm (o mg/L). Se encontró que en 55 personas sanas de ambos sexos el NCBC se halló en concentraciones muy bajas siendo el X de 0,95±0,10 ppm lo cual está de acuerdo por lo informado en la literatura específica (26).
Metabolismo lipídico y lipoproteico. Los parámetros convencionales (CT: colesterol total, C-HDL: colesterol transportado por proteínas de alta densidad o HDL, C-LDL: colesterol transportando por proteínas de baja densidad o LDL y TAG: triglicéridos plasmáticos) se midieron usando equipos comerciales de la firma Wiener Lab. (Rosario, Argentina). Los resultados se expresaron en mg/dL. Las apolipoproteínas se determinaron mediante equipos comerciales ad-hoc: Apo-B100 ELISA Kit (ALP) de Mabtech. Inc. (OH, EE.UU.), Apo-D se midió con el Human Apolipoprotein D ELISA Kit de MyBioSource (NY, EE.UU.) (ambas con un rango de detección de 3 a 30 mg/dL). La Apo-A1 se determinó con el SPQTM II Kit 86070 Liquid, de DiaSoria S.A. (Madrid, España) de tipo nefelométrico. La LDL-oxidada (Ox-LDL) se determinó mediante un ensayo ELISA sandwich con anticuerpos monoclonales dirigidos contra determinantes antigénicos de la Apo-B modificados por estrés oxidativo, el cual se adquirió como [10007893-OxLDL-b2GPI] (Human) ELISA Kit a la empresa Cayman Chem. Co. (MI, EE.UU.). Los resultados se expresaron como el cociente Ox-LDL/LDL tal como lo recomienda Galland et al. (132). La Lp(a) se midió por turbidimetría con el equipo comercial DiaSorin (Sallugia, Italia) calibrado con los estándares incluidos por el propio fabricante. Ninguno de los participantes fue portador homocigota del polimorfismo ApoE4 determinado mediante la técnica de PCR con el equipo comercial LightMix Kit ApoE C112R R158C® de Roche, (Paris, Francia) comercializado por TIB-MolBiol bajo el Cat.-No. 40-0445-16.
Relación entre glutatión oxidado (GSSG) y reducido (GSH). El cociente GSSG/GSH se determinó midiendo el glutatión total y luego el reducido. El oxidado se obtuvo por sustracción. Para la determinación se siguió la metodología de Anderson y Meister (133). En esta técnica se expuso la muestra a cantidades excesivas de ácido 5,5-ditiobis-2-nitrobenzoico (DTNB) para transformar todo el GSH presente en GSSG. En una segunda fase de la reacción el GSSG se redujo a GSH por una reacción acoplada catalizada por la glutatión reductasa NADPH-dependiente. A medida que la enzima produjo GSH éste reaccionó con el DTNB remanente generando ácido 5-tio-2-nitrobenzoico (TNB). La velocidad de aparición de TNB se pudo monitorear a 412 nm, y resultó directamente proporcional a la concentración de glutatión en la muestra. Para realizar el análisis se agregaron 500 μL de buffer fosfato sódico 140 mM/EDTA 6,3 mM (pH: 7,50); 0,7 mL de la solución de NADPH (0,25 mg/mL); 283 μL de la mezcla de DTNB 6 mM en ácido tricloroacético (TCA) al 5% y la alícuota de plasma (100 μL). Se incubó a 30 oC por 10 minutos. Luego se agregaron 2,8 μL de una solución que contenía 5 UI de la enzima glutatión reductasa (Sigma Chem. Co., EE.UU.). Se mezcló por inversión y se colocó inmediatamente en el compartimiento del espectrofotómetro de doble haz Cintra-20 (Sydney, Australia) termostatizado a 25 oC y se registró la cinética de la reacción durante un período de 2 minutos. Para realizar los cálculos de substracción se repitieron los pasos anteriores pero agregando 3 mM de divinil-piridina que bloquea al GSSG. La calibración se efectuó utilizando varias alícuotas de una solución testigo de GSH en buffer fosfato sódico (con 2/3 del volumen en solución de TCA al 5%).
Capacidad antioxidante total del plasma (ensayo FRAP). Se siguió la conocida metodología de Benzie y Strain (134). A pH ácido se puede determinar la habilidad de cualquier líquido biológico (plasma por ejemplo) para reducir iones férricos a ferrosos. Esto se puede cuantificar colorimétricamente por formación de un complejo entre el hierro ferroso y la tripiridil-triacina (TPZ) mediante la formación de un cromóforo de intenso color azul con un pico de absorción a los 583 nm. Se trata de una reacción no específica que tiene en cuenta a todos los componentes con capacidad reductora (bilirrubina, ácido úrico, vitámeros, etc.). En este caso, el ensayo valoró la totalidad de las sustancias antioxidantes circulantes en el plasma excepto el ácido úrico el cual fue deliberadamente destruido por incubación previa con uricasa (Sigma Chem. Co, Buenos Aires) agregada en exceso y disuelta en buffer fosfato 300 mM/glicerol 20 mM (pH 7,40) a razón de 30 UI por cada 500 μL de muestra. Esto fue seguido de incubación a 37 oC por 15 min en un agitador orbital. Así se evitó la dispersión de los resultados inherentes al disímil contenido de ácido úrico entre los integrantes del estudio y permitió referirse solamente a los antioxidantes endógenos biológicamente activos de las muestras. Para hacer el ensayo FRAP se tomaron 3 mL de la solución reactivo de trabajo compuesta por buffer acético/acetato 300 mM (pH 3,60), solución de TPZ 10 mM (Sigma Chem. Co, EE.UU.) en HCl 40 mM y solución de cloruro férrico 20 mM (Mallinckrodt, EE.UU.) mezcladas en proporción 83: 8,5: 8,5; respectivamente. A 3 mL de este reactivo colocado en una cubeta de 1 cm de paso de luz y termostatizada a 25 oC, se le agregaron 90 μL de plasma pre-tratado con uricasa. Tras la mezcla inmediata por inversión se obtuvieron los datos de DO (583 nm) vs tiempo durante 4 minutos mediante un registro computacional continuo. De cada curva se calculó la pendiente (tangente de la curva a tiempo cero o velocidad inicial de reducción) de incremento de la densidad óptica y se la comparó con la pendiente obtenida para un plasma estándar (Sigma Chem. Co, Buenos Aires) cuya capacidad antioxidante resultó conocida. A su vez, el valor de FRAP de esta muestra de referencia se constató (al solo efecto de efectuar control de calidad) por calibración con una solución comercial de concentración conocida de Trolox® (diacetato de α-tocoferol) de Fluka (Berlín, Alemania).
Carbonilos proteicos (CPs). Se realizó la técnica descripta por Reznick y Packer (135) que es de aceptación internacional por la reproducibilidad de sus resultados. Esta metodología detecta la formación de grupos carbonilos u "oxo" que se forman en las proteínas debido a la oxidación de los aminoácidos de las cadenas laterales por las especies radicalarias de cualquier naturaleza (ROS y RNS). Estos carbonilos reaccionan con la 2,4-dinitrofenilhidracina (2,4-DNPH) para formar dinitro-fenilhidrazonas de color caoba que pudieron ser cuantificadas por espectrofotometría. Las alícuotas de las muestras (80 μL) se llevaron a volumen final de 1,5 mL con solución buffer fosfato pH 7,40 (PBS). Se agregaron 1,5 mL de DNPH 10 mM en HCl 2 N y se incubaron en oscuridad durante 30 minutos a temperatura ambiente. Al finalizar la incubación, se le agregó a cada tubo 2 mL de una solución de NaOH 2 N y 1 mL de etanol absoluto para evitar la turbidez debida a restos de proteína desnaturalizada. Luego de cada agregado se agitó de inmediato y vigorosamente. De esta manera se obtuvieron complejos de color caoba que se leyeron a 505 nm en el espectrofotómetro de doble haz Cintra-20. La curva de calibración se efectuó utilizando una solución estándar de piruvato de sodio 2,30 mM mediante alícuotas comprendidas dentro del rango 40 a 100 μL cuyas concentraciones estuvieron ubicadas por debajo y por encima de los valores esperados para las muestras de los integrantes del estudio.
Sustancias reactivas al ácido tiobarbitúrico (TBARS). El método empleado fue sensible y altamente reproducible y se encuentra descripto por Yagi (136). Esta técnica se basó en la reacción del ácido tiobarbitúrico con productos secundarios de la peroxidación lipídica, tales como el malondialdehído (MDA), 4-hidroxihexanal y 4-hidroxinonenal, para formar aductos de color rosado que pueden ser leídos en un espectrofotómetro a una densidad óptica de 532 nm. Se tomaron alícuotas de 50 μL de plasma que se diluyeron con PBS a un volumen final de 200 μL. Luego se adicionaron 200 μL de SDS (dodecilsulafo sódico) (Sigma Chem. Co., EE.UU.) al 8,1% y se agitó teniendo cuidado de no hacer excesiva espuma. A continuación se añadieron 1,5 mL de buffer acético/acetato 100 mM (pH 3,50); 1,5 mL de TBA al 0,8% y 600 μL de agua bidestilada. Las muestras se incubaron a 95 oC durante 60 min. Pasado este tiempo se dejaron enfriar y se les agregó 1 mL de agua bidestilada y 3 mL de alcohol isobutílico agitando vigorosamente. La fase orgánica se leyó en el espectrofotómetro Cintra-20 a una longitud de onda de 532 nm. Para determinar las concentraciones de TBARS en las muestras problema se realizó una curva de calibración utilizando como testigo una solución comercial de MDA (bis-dimetil-acetal o 1,1,3’,3’-tetrametoxipropano) en el rango de concentraciones 4,5 a 36,0 nmoles de MDA/tubo.
Determinación de la concentración de péptidos beta-amiloides. Los péptidos bA1-40 y bA1-42 se determinaron en sangre periférica utilizando el equipo comercial Human/Rat B Amyloid (1-42) ELISA Kit Wako I (CA, EE.UU.), High-Sensitive [BNT77/BC05 (Fab’)] Cat. # 296-64401 con un rango de detección lineal: 0,1–20,0 pM y Human/Rat B Amyloid (1-40) ELISA Kit Wako II (CA, EE.UU.) [BNT77/BA27(Fab’)2] Cat. # 294-64701 con un rango de detección lineal: 1–100 pM. Justo antes de ser analizadas, las muestras se centrifugaron a 6.000 g por 15 min a 2 oC y luego se diluyeron al doble con buffer de dilución provisto por el fabricante del equipo. Se usaron alícuotas de plasma de 15 μL (de dilución)/ tubo. Cada paciente fue analizado por triplicado y los resultados se promediaron y se expresaron en concentración pM.
Proteínas totales. Se utilizó el método de Bradford (137) que es rápido, sensible, lineal y muy simple de llevar a cabo. De alta reproducibilidad y color final estable por una hora (con solo un 4% de decrecimiento de la densidad óptica) es apropiado para muestras con alto contenido proteico como las analizadas en este estudio. Se basa en el cambio de absorbancia que experimenta el colorante Brillant Coomassie Blue G-250 (BCB-G250) de Fluka (Berlín, Alemania) al ligar proteínas. El máximo de absorbancia ocurre a 595 nm. Como patrón de calibración se empleó una solución de albúmina pura (Sigma Chem. Co., Buenos Aires) en PBS de 7,0 g/dL.
Evaluación cognitiva. Se llevó a cabo la evaluación cognitiva de todos los integrantes del estudio (controles y pacientes) mediante el test MMSE que es sencillo, reproducible y estandarizable, además de correlacionar muy bien con baterías de evaluación complejas. Se empleó una versión de la prueba adaptada a nuestra población (con línea de corte 24) y se tuvo en cuenta la ponderación de los puntajes de cada área por la escolaridad/formación académica del paciente o control. En ambos grupos no hubo analfabetos y todos tuvieron –al menos– 6 años de escolaridad. El MMSE o mini examen del estado mental (Mini Mental State Examination, en inglés) en versión original de Folstein et al. (115) se implementó en español rioplatense según Allegri et al. (138), revisada por Butman et al. (139). La implementación siguió al Grupo de Neuropsicología Clínica de la Sociedad de Neurología de Argentina o SNA (140), y también las recomendaciones de Burin et al. (141) y de la Revista Neurológica Argentina (142).
Aspectos bioéticos y legales. El protocolo se consideró "no-invasivo" y se ajustó a la Declaración de Helsinki (http://www.bioetica.uchile.cl/doc/helsinki.htm) según el documento actualizado a partir de la primera declaración de la Asociación Médica Mundial (Declaración de Helsinki, Finlandia; 1964). Los aspectos legales estuvieron basados en el modelo descripto en la URL http://www.easp.es/legislacion/real+decreto+561+. htm. El procedimiento estuvo en un todo de acuerdo con lo establecido por el Comité de ética del CONICET en sus documentos regulatorios (143), y con lo establecido por la OMS (Organización Mundial de la Salud) (144). Asimismo, el estudio se sometió a evaluación independiente por el Honorable Comité de Bioética de la Facultad de Ciencias Médicas de la Universidad Nacional de La Plata (COBIMED) que lo aprobó sin objeciones bajo el registro protocolar # 0800-002982- 12/00 y del Comité de Bioética del Hospital de Gonnet que lo aprobó por nota de fecha 10-06-2012 con inclusión de la carta-compromiso que establece la ley Nac. Nº 25326 en el formulario del consentimiento y que se refiere a lo establecido por la Dirección Nacional de Protección de Datos Personales para el legítimo resguardo de la confidencialidad (de acuerdo con la regulatoria y medidas de implementación de la jurisprudencia vigente).
Bioestadística. Excepto que se explicite otro valor, la significación establecida fue 0,01<p<0,05. En general los datos se expresaron como el promedio (X) ± una desviación estándar (DE) de la media en forma absoluta para los resultados de los análisis de referencia (C y EA). Sin embargo, para expresar algunos resultados en forma comparativa se recurrió a la expresión de porcentaje de cambio respecto a su correspondiente control. En esta modalidad cada uno de los datos (X´) se calculó en términos de porcentaje de cambio (PCRC) respecto a ese mismo parámetro en el grupo que ofició como referencia (R) (145), siendo: PCRC (X´) = [X–R].100/R. Para el análisis de los resultados de MMSE en algunos estudios de regresión y correlación también se utilizaron datos normalizados por DE. La normalización se llevó a cabo mediante la expresión: Pe = [P-Ppg]/DEpg la cual fue tomada de Cunqueiro et al. (146). En este cálculo Pe es la variable estandarizada que se pretendió normalizar para cada paciente-caso; P es la variable bruta; Ppg es la variable media dentro de su correspondiente grupo experimental y DEpg es la desviación estándar de la variable para esa población. Por esto, la puntuación media resultante para cada grupo (promediada a partir de los Pe) estuvo expresada en unidades de DE. Esta normalización se empleó específicamente para correlacionar una función definida como "FAE" (Función de Adición de Efectos) que será explicada en la parte de resultados experimentales.
Resultados
Aspectos demográficos de la población estudiada y características generales de cada subgrupo. Tal como antes se mencionó, al grupo de personas controles se lo designó con la letra C, mientras que los subgrupos de pacientes con enfermedad de Alzheimer se denominaron L-EA, I-EA y S-EA. Cuando se haga alusión al grupo EA (sin prefijo alguno) se estará refiriendo al total de pacientes con enfermedad de Alzheimer sin distinción del estadio clínico. Entre los subgrupos –y entre estos y el grupo control– no hubo diferencias estadísticamente significativas en el promedio de los años de educación recibida, estándar socioeconómico, estado marital (porcentaje de casados y/o viviendo con pareja estable) ni en el número de hijos concebidos por pareja (2 en promedio; datos no mostrados). No se registraron diferencias significativas en la mayoría de los aspectos encuestados entre las poblaciones de pacientes EA y los sujetos empleados como controles con excepción (obviamente) de las indicaciones farmacológicas. Una encuesta nutricional retrospectiva basada en los lineamientos establecidos por el Ministerio de Salud de la Nación Argentina demostró diferencias significativas entre controles y pacientes atribuibles muy probablemente a que el grupo EA posee un espectro más restringido de selección de alimentos (en la mayoría de los casos por carecer de la iniciativa de decidir sus menús). Estas diferencias se refirieron al tipo de alimento antes que a su clase general. Sin embargo, este hecho no significó diferencias en cuanto al balance de nutrientes esenciales ni a la ingesta calórica recomendada para cada índice de masa corporal (IMC). Hubo una significativa diferencia en los consumos de alcohol y de grasas saturadas que fueron menores en el grupo EA (de cualquier subcategoría) que en los C. Por esta razón en el análisis de regresión múltiple destinado a comparar grupos entre sí, se consideró a las calorías-día estimadas a partir de alcohol y de las grasas saturadas como distractor para la significación de las diferencias en cualquiera de los datos contrastados. Otros comentarios específicos sobre la encuesta nutricional se harán más adelante. Algunos valores relevantes –recopilados para las características demográficas y generales de los subgrupos estudiados– aparecen como información fraccionada en las (Tabla I) (Tabla II)(Tabla III).
Tabla I. Características demográficas y generales para los subgrupos integrantes del estudio
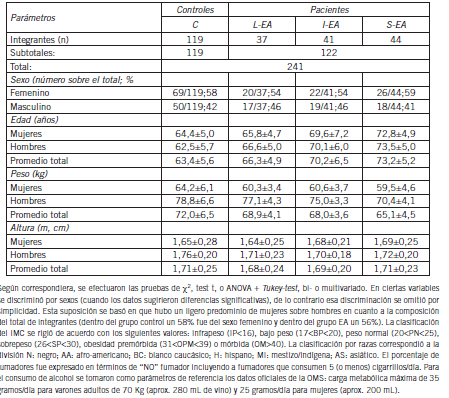
Tabla II. Características demográficas y generales para los subgrupos integrantes del estudio (continuación)
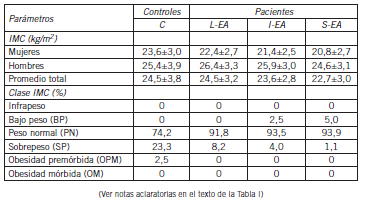
Tabla III. Características demográficas y generales para los subgrupos integrantes del estudio (continuación)
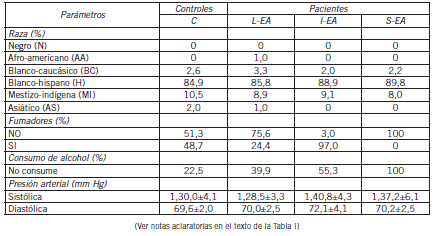
La composición racial de los subgrupos no fue significativamente diferente. El consumo de tabaco y de alcohol fue significativamente menor (p<0,01) en los pacientes y dentro de ellos decreció con el progreso de la enfermedad (p<0,05). El peso corporal y la distribución en categorías según el IMC fueron también diferentes según el sexo (p<0,05). Solo se observaron casos de bajo peso en los subgrupos I-EA y S-EA: 1 caso de 41 (2,5%) y 2 casos de 39 (5%); respectivamente.
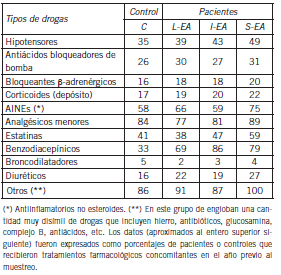
Es importante notar que los esquemas de tratamiento farmacológico de los pacientes coincidieron en muchos aspectos pero no en otros (tales como el tipo de droga y la posología). Algo similar sucedió con la duración de los esquemas y su recambio durante la evolución clínica del paciente. Además, tanto pacientes como controles recibieron medicación (ocasional o sistemáticamente) por eventos esporádicos o intercurrencias. Esto hizo imposible sistematizarlos como variables a considerar en los estadísticos de prueba. A modo solamente informativo se muestran las drogas empleadas como base terapéutica en el tratamiento de los pacientes EA y otras drogas coadyuvantes (Tabla IV).
Tabla IV. Tratamientos farmacológicos de pacientes con EA (expresados como porcentajes de pacientes que reciben la droga al momento del muestreo)
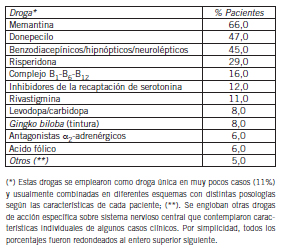
Se puede ver que casi el 70% de los pacientes recibió donepecilo, solo o combinado con memantina, y un grupo menor se trató con rivastigmina. Casi la mitad de los pacientes recibió algún tipo de droga benzodiacepínica, hipnótica, o alguna otra clase de neuroléptico. El 30% (aproximadamente) fue tratado con risperidona. Asimismo, del examen de las historias clínicas se obtuvo la información sobre los tratamientos farmacológicos concomitantes con la patología de base, muchos de los cuales se implementaron como consecuencia de la evolución clínica del propio cuadro, o por otras causas, tanto para el grupo C como para el EA. Estos datos aparecen detallados en la Tabla V.
Tabla V. Tratamientos farmacológicos concomitantes durante el año previo a la toma de muestra
Para concluir con la parte descriptiva de la población, se resumen en la Tabla VI los eventos concomitantes sobresalientes de sus historias clínicas. Algunos datos no presentaron un patrón claro; sin embargo, en otros casos se hizo evidente que los pacientes tuvieron mayor cantidad de episodios de infección de vías aéreas (y otras infecciones en general) y que esto se acrecentó con la gravedad del cuadro clínico de base. También es evidente que los pacientes presentaron mayor cantidad de casos de dislipemias y de coronariopatías, como así también algún tipo de distimia asociada a la propia EA. También es probable que haya un mayor número de casos de hipoacusia entre los pacientes, pero esto es frecuentemente muy difícil de evaluar en personas dementes por lo que este dato debe tomarse con cautela. Al comparar otras patologías entre C y EA se obtuvieron incidencias similares.
Tabla VI. Principales cuadros clínicos concomitantes (en porcentajes de incidencia dentro de cada subgrupo) ocurridos durante el último año

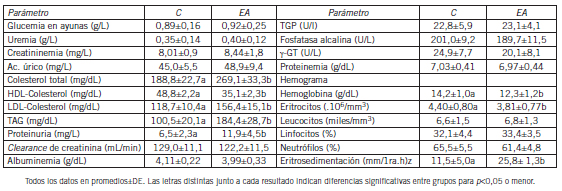
Bioquímica clínica. Seguidamente se muestran los datos de química clínica basal para los controles y subgrupos estudiados considerados en su conjunto (hombres y mujeres) (Tabla VII). A los fines de simplificar la presentación de los datos se muestran solo los promedios de los grupos C y EA. Las diferencias entre sexos como así también las posibles diferencias dentro de cada sub-grupo EA se comentarán en forma simultánea. La mayoría de los resultados no presentó sesgo por el sexo con excepción de los valores de g-glutamiltranspeptidasa (g- GT) (p<0,05) que fueron ligeramente más altos en los hombres que en las mujeres mostrando un incremento de tipo fisiológico (sin significación clínica pues se lo atribuyó a la mayor masa muscular). En el promedio general de cada grupo (C y EA) esta diferencia no se vio reflejada. Hubo una tendencia estadísticamente no significativa a una mayor trigliceridemia en las mujeres tanto del grupo C como de cualquiera de los subgrupos EA que fue muy discreta y estuvo en el orden del 15 al 19% (mayor que los valores promedio de hombres en el correspondiente subgrupo). A su vez, los valores promedio (para el total de integrantes de cada subgrupo) de TAG fueron más altos (p<0,01) para los pacientes EA que para el grupo C. El colesterol total mostró diferencias entre grupos con mayores valores dentro del grupo EA que en el control (p<0,05). El colesterol transportado por HDL fue menor (p<0,05) para el grupo EA con un progresivo y significativo declive dependiente del empeoramiento del cuadro clínico (se lo mostrará en detalle más adelante). A pesar de que normalmente este parámetro es menor en hombres que en mujeres, esto no se observó en este estudio probablemente por tratarse de personas de edad avanzada integrado por mujeres post-menopáusicas. En los promedios generales el colesterol LDL fue mayor (p<0,01) para el grupo EA comparado con el control y también mostró una tendencia alcista con el progreso del cuadro clínico dentro del grupo de los pacientes. La proteinuria fue también más alta en el grupo EA (p<0,05) sin que este parámetro tuviera diferencias ligadas al sexo. Sin embargo, los valores de uremia y de depuración de creatinina endógena (como biomarcadores de la función renal) fueron indistinguibles desde el punto de vista estadístico. Los otros datos no merecen consideraciones especiales con excepción de una disminución de algunos valores hematimétricos (eritrocitos y hemoglobina) dentro del grupo EA que también mostraron una disminución significativa con el empeoramiento del cuadro clínico y sin asociación con el sexo (de hecho, los L-EA no presentaron diferencia con los controles pero los I-EA y S-EA mostraron disminuciones progresivamente significativas). La eritrosedimentación (primera hora) fue significativamente más elevada (p<0,05) en el grupo EA comparado con el C. Otros valores que no son de determinación rutinaria se comentarán en una sección aparte.
Tabla VII. Resultados de análisis de química clínica básica en los grupos experimentales
Homeostasis del cobre: marcadores periféricos. Los datos concernientes específicamente al metabolismo del cobre que se pueden valorar en sangre periférica aparecen en la Tabla VIII.
Tabla VIII. Biomarcadores periféricos del metabolismo del cobre

Se observó un comportamiento diferencial entre las dos proteínas circulantes a nivel periférico que están vinculadas al metabolismo del cobre. Por un lado, las metalotioneínas (MTs) se mantuvieron sin diferencias significativas entre C y EA ni tampoco entre los subgrupos de EA. Por otra parte, la ceruloplasmina (CRP) se halló más elevada en los pacientes leves e intermedios de EA pero fue de un valor indistinguible al del control en el grupo EA severo. A su vez, los pacientes I-EA tuvieron menos CRP que los L-EA, sugiriendo un incremento en los cuadros más leves que con el tiempo de evolución declinó hasta alcanzar los valores del grupo control. La cupremia (Cu total), en cambio, se incrementó en todos los grupos EA a medida que el cuadro progresó en comparación con los valores registrados para el grupo control. Lo mismo sucedió con el cobre libre no unido a ceruloplasmina (NCBC) siendo en este caso mucho más notable el incremento registrado en función de la severidad del cuadro. Estos cambios quedaron reflejados en los cocientes entre Cu total o NCBC y las proteínas MTs y CRP que mostraron un incremento en los casos más avanzados de EA respecto al valor control. Aún más significativo fue el aumento del cociente NCBC/Cu total que representa a la fracción de la cupremia total no complejada a proteína.
Cobre en alimentos y en agua de bebida. Por medio de la encuesta nutricional estandarizada retrospectiva se estimó el total de cobre ingerido por día y por integrante, y asimismo se calculó la cantidad de cobre orgánico e inorgánico basándose en los datos proporcionados por la OMS para América Latina y el Caribe (http://www.scielo.sa.cr/pdf/rcsp/v21n2/art09v21n2.pdf). Los resultados se muestran en la Tabla IX.
Tabla IX. Estimación de los contenidos de cobre ingeridos
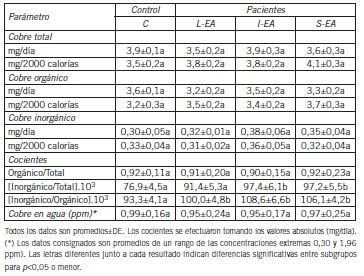
Se distinguió entre cobre inorgánico y orgánico para referirse a las sales minerales de este metal de transición (disueltas o en suspensión coloidal) o a los compuestos complejos (péptidos, proteínas, y otras sustancias orgánicas de estructura variada que contienen al metal coordinado o unido covalentemente) pues se sabe que su papel es distinto en cuanto a su potencialidad tóxica y capacidad pro-oxidante. Las formas químicas inorgánicas del cobre comprenden varias especies moleculares (óxidos, hidróxidos, oxicloruros, sulfatos y carbonatos principalmente). El cobre total ingerido por día/integrante fue indistinguible entre grupos. Cuando se ponderó esa ci fra en relación a las calorías-día (estimadas por encuesta) tampoco aparecieron diferencias entre subgrupos. Este cálculo se efectuó porque las calorías-día no fueron iguales para todos los integrantes del estudio (en promedios±DE: 2,200±145 para C, 1,860±121 para L-EA, 1,799 para I-EA y 1,720±102 para S-EA; todas las dietas estuvieron comprendidas entre 25 y 30 calorías/kg de peso (o kcal). En relación al subtipo de cobre adquirido por medio de la alimentación y en comparación al del agua de bebida y uso para cocción, la proporción ingerida de cobre orgánico en relación al total fue la misma para todos los subgrupos, pero no así la de cobre inorgánico que mostró valores ligeramente más altos (p<0,05) en los subgrupos EA respecto a los C. Asimismo, el promedio de la concentración de cobre en agua de bebida fue idéntico entre los subgrupos a pesar de que varios integrantes utilizaron fuentes de provisión de agua distintas (agua de red o de pozo). Este parámetro merece un comentario adicional pues los valores dependieron fuertemente de la región o localización urbana del participante y se movieron entre rangos muy amplios comprendidos entre 0,30 y 1,96 ppm. Esto quedó demostrado por el valor comparativamente elevado de la DE de los datos (entre 18 y 26% de la media). En relación a esta última cuestión se efectuó un relevamiento del nivel de cobre en la zona del Gran La Plata mediante un muestreo en condiciones idénticas a las implementadas durante el estudio (toma purgada). Si se tiene en cuenta que el total de cobre inorgánico ingerido por día/integrante fue de aproximadamente 0,35 mg/día y que un litro de agua puede tener entre 0,30 y 1,96 mg, el papel desempeñado por la ingesta de agua sobre esta variable se torna absolutamente relevante. El estudio se muestra en la Figura 1 en donde se graficaron los resultados de este relevamiento en formas de curvas de nivel que delimitan tres zonas más o menos definidas (una de ellas interrumpida por otra en dos sectores). Existe un área de menor concentración promedio (0,51±0,12 ppm, en verde), una de valores intermedios (0,92±0,23 ppm, en amarillo) y otra de valores muy altos (1,65±0,27 ppm, en rosado). La última delimita una franja que se dirige en dirección norte-sur y coincide con la denominada "cuenca del arroyo El Gato".

Figura 1. Distribución asimétrica de la concentración de cobre en agua de consumo en el Gran La Plata
Los datos que se tomaron corresponden al promedio de dos análisis de agua de la misma vivienda separados una semana entre sí. Fueron procesados y graficados para curvas de nivel por el software de Erdas v.11.1 y ajustados a tres zonas de valores promedio: bajo, intermedio y alto.
De estos resultados surgió que podría haber una relación o -correlación estadísticamente significativa- entre la zona de residencia y alguno de los parámetros investigados en este estudio. Lamentablemente, la encuesta nutricional retrospectiva arrojó datos poco confiables sobre la cantidad de agua que cada participante ingirió por día (muy a pesar de haber sido modificada ex profeso para ser más precisa respecto de este parámetro). Esta cuestión impidió calcular de manera confiable el volumen-día y por consiguiente la ingesta de cobre inorgánico a partir de esta fuente, pero por lo antes considerado y teniendo en cuenta que aproximadamente cada persona ingirió un litro de agua/día, la cantidad de cobre adquirida por este medio debió ser considerable (1 mg o más/24 h).
Biomarcadores de estrés y daño pro-oxidativo. A continuación se muestran (Tabla X) algunos biomarcadores no convencionales o emergentes determinados en plasma con el objeto de ponderar el impacto del estrés oxidativo sobre lípidos y proteínas en C y EA.
Tabla X. Estado redox y biomarcadores de daño pro-oxidativo en plasma

Los resultados concuerdan con un escenario de estrés oxidativo y daño pro-oxidativo en la enfermedad de Alzheimer. Se observó un incremento de la peroxidación lipídica demostrada por la elevación progresiva de los TBARS conforme se agrava el cuadro clínico y una mayor carbonilación proteica (elevación de los PCs). Concomitantemente, descendió la capacidad antioxidante total del plasma (ensayo FRAP) lo cual demostró un agotamiento del sistema de defensa antioxidante no enzimático. Esto último estuvo acompañado por una congruente disminución del glutatión reducido y un incremento del oxidado (con la consiguiente elevación progresiva del cociente GSSG/GSH).
Perfil lipoproteico y marcadores de riesgo para enfermedad vascular. En la Tabla XI aparecen los resultados de las determinaciones de apolipoproteínas y el cálculo de varios marcadores de riesgo vascular. De los resultados mostrados en esta Tabla se desprende claramente que toda la población EA presentó elevación significativa de muchos de los indicadores de riesgo aterogénico con independencia del estadio clínico de la patología. Esto concordó con estimadores groseros del riesgo vascular proyectado a 10 años de evolución.
Tabla XI. Perfil lipoproteico en plasma

Se observó que ni el colesterol total (CT) ni ninguna de sus fracciones (HDL-C, LDL-C) o los triglicéridos (TAG) mostraron una relación clara con el estado evolutivo de la enfermedad de Alzheimer. Sin embargo, todos esos parámetros estuvieron significativamente más elevados en los pacientes comparados con los controles (p<0,05 o menor). Mucho más interesante fue el comportamiento de la fracción peroxidada de HDL que se incrementó en los I-EA y S-EA respecto de los C y este incremento se vio claramente ligado al estadio de la enfermedad. Esto último se reflejó en el cociente Ox-LDL/LDL. Asimismo, la proporción entre TAG o CT y HDL-C fue más alta en los pacientes que en los controles aunque nuevamente no se observó una clara dependencia con el estadio clínico. Todos estos parámetros significaron un progresivo aumento del riesgo de enfermedad vascular para los pacientes EA reflejado en un aumento con el agravamiento de la patología de base. Por el contrario, la Lp(a) (lipoproteína "a") que se emplea como marcador pro-aterogénico se encontró claramente incrementada en los pacientes EA con una visible dependencia con el estadio evolutivo. Las apolipoproteínas D, B y A1 correlacionaron con los valores de HDL colesterol (D y A1) y LDL colesterol (B), mientras que las relaciones entre Apo-B100/Apo-A1 y Apo-B100/Apo-D mostraron significativos incrementos en los sub-grupos EA comparados con los controles.
Evaluación cognitiva. Se llevó a cabo la evaluación cognitiva mediante el test MMSE (como ya se aclaró en una versión validada para español rioplatense) de todos los integrantes del estudio (C y EA) en un entorno controlado o estandarizado, en silencio y sin intrusiones. Esta prueba puntúa como máximo 30 y los ítems están agrupados en 5 apartados que consideran orientación, memoria inmediata, atención y cálculo, recuerdo diferido, lenguaje/simbología y construcción. Para la comprobación de la memoria inmediata (o memoria de fijación o de trabajo) cada pausa fue de 1 segundo, y se le repitió la serie hasta 5 veces aunque en este apartado solo se registraron las respuestas obtenidas tras la primera serie. La intención de las repeticiones (las que fueron necesarias, pero hasta el límite referido) fue que el participante las aprendiera para la subsiguiente evaluación en el apartado de memoria diferida. En el área de lenguaje y construcción, se comprobó la posible presencia de trastornos afásicos, de apraxia ideatoria, de apraxia de construcción, etc. Se consideró un valor de corte "promedio" de 24 puntos; sin embargo, se debe aclarar que en cada caso se tuvo en cuenta el nivel de instrucción y se empleó una Tabla de ajuste como la que aparece a continuación (Tabla XII) y que es una adaptación hecha por Crum et al. (147).
Tabla XII. Tabla de ponderación para el MMSE

Tabla XIII. Evaluación cognitiva por medio del MMSE

Los resultados indicaron diferencias muy significativas (p<0,01) respecto al control para cada estadio clínico del EA. Existió muy escasa superposición de valores al cambiar de subgrupo (lo cual concuerda con las bajas medidas de dispersión de los datos en torno a la media). El cociente EA/C reflejó el deterioro relativo de la variable MMSE para cada subgrupo, mientras que el porcentaje de cambio respecto al control (PCRC) indicó un progresivo empeoramiento de la variable estudiada a medida que se agravó el cuadro clínico. Es importante consignar que dentro del subgrupo clasificado como L-EA con un MMSE de 20,0±2,3 se incluyeron algunos pacientes con deterioro cognitivo leve (DCL) que no está claro aún si se trata de pacientes que evolucionarán (o no) a casos de EA propiamente dichos. Este grupo de pacientes con queja cognitiva tuvieron puntajes dentro del rango 23-26 y constituyó el 24% del total dentro de su categoría (9 de 37). Por otra parte se determinó el contenido de los péptidos bA en plasma periférico midiéndose por separado los fragmentos 1-40 y 1-42, encontrándose los resultados que se indican en la Tabla XIV.
Tabla XIV. Evaluación de la concentración plasmática de bA

Según lo detallado por el fabricante del equipo comercial los datos (expresados en concentración pM) deberían esperarse en valores promedio de 5,25 y 58,47 para los fragmentos bA1-40 y bA1-42, respectivamente. El método tiene una detección lineal entre 1 y 100 pM, con casi 100% de especificidad y aproximadamente 95% de recuperación. Se observó que en la población control los resultados obtenidos se hallaron dentro de los rangos esperados. Sin embargo, hubo un incremento significativo del bA1-42 que acompañó al agravamiento del cuadro clínico. No sucedió lo mismo con bA1-40 que únicamente tuvo valores elevados para el subgrupo que padecía EA severo. El cociente bA(1-40)/(1-42) también se incrementó paulatina y significativamente a medida que los pacientes empeoraron su condición clínica. Esto último quedó fielmente reflejado en los porcentajes de cambio calculados para cada subgrupo respecto del control.
Estudios de correlación y regresión. Se llevaron a cabo estudios de correlación y de regresión entre variables seleccionadas para investigar en primer término si existía una correlación entre los valores de algunos de estos biomarcadores entre sí y en segundo lugar para determinar si los valores de la variable MMSE correlacionaban particularmente de manera significativa con el colesterol, el nivel de cobre y/o la concentración de péptidos bA. En algunos casos se efectuaron ajustes y ponderaciones que se indicarán en cada caso. Antes de considerar estos resultados, cabe consignar que mediante análisis por ANOVA-Tukey (como test post-hoc) se constató que las diferencias entre el nivel de cobre en agua de bebida/uso alimenticio fueron significativas (p<0,01) para cualquier par de comparaciones en las tres zonas urbanas delimitadas por el muestreo. En este estudio hubo tomas correspondientes a integrantes del estudio y otras adicionales de no integrantes. Considerando solo a los integrantes, no fue factible establecer una ponderación confiable por el tiempo de habitación dentro de cada zona, lo cual hubiera sido muy interesante desde el punto de vista de la influencia de esta variable (cobre en agua) sobre los niveles de Cu, NCBC o cuproproteínas en sangre periférica. Sin embargo, sí se pudo establecer que el 100% de las muestras correspondientes a integrantes representaron más de un año (el inmediatamente anterior al estudio) de permanencia dentro de una zona en cuestión. Un 83% correspondió a más de 10 años, 11% al intervalo 5-10 años, y el resto (6%) a un periodo mayor a 1 pero menor a 5 años. Si se toman las otras variables continuas que se determinaron y se las pondera o ajusta por la duración del cuadro clínico, es posible efectuar una multicorrelación entre ellas (damero de Pearson), lo cual resultó en los datos mostrados en las Tablas XV y XVI, que estudiaron los posibles vínculos –entre el cambio de pares de variables que se considere– para: (a) la concentración de cobre/ cuproteínas vs el nivel de estrés oxidativo/daño oxidativo en el primer caso; y (b) los niveles de cobre vs los biomarcadores del metabolismo lipoproteico en el segundo caso. Los datos significativos se indicaron con un asterisco. Por simplicidad no se consignaron los niveles de significación pero en todos los casos fueron ≤0,05.
Tabla XV. Correlación entre pares de variables seleccionadas (preajustadas por tiempo de evolución) para pacientes en relación al daño pro-oxidativo y nivel de cobre/cuproproteínas
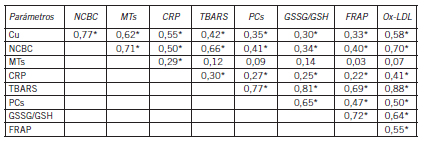
Tabla XVI. Correlación entre pares de variables seleccionadas (preajustadas por tiempo de evolución) para pacientes en relación al metabolismo lipoproteico y nivel de cobre
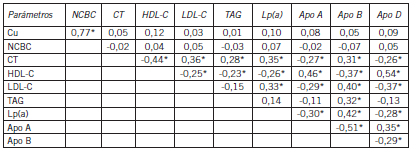
Se puede ver claramente que existió un vínculo evidente entre el nivel de Cu y especialmente el de NCBC (los coeficientes son más cercanos a la unidad comparados con los de Cu) y las variables que indicaron estrés o daño oxidativo. El valor del NCBC estuvo en correlación positiva con la cupremia total. También correlacionaron entre si y en forma positiva las dos cuproproteínas analizadas (MTs y CRP).
Es interesante observar que los parámetros del perfil lipídico y apolipoproteico no guardaron ninguna correlación evidente con los valores de cupremia total ni con el de cobre libre. Este resultado sugirió que no son variables que se influencien mutuamente y deberían considerarse como independientes. Existieron otras correlaciones entre marcadores del metabolismo lipídico que reafirmaron conocimientos previos, tales como el vínculo inverso entre colesterolemia total y HDL-C o el directo con LDL-C. A su vez HDL-C y LDL-C mostraron una correlación inversa entre ellas. En cuanto a las apolipoproteínas, se observó claramente que hubo una correlación positiva entre Apo D o Apo A y HDL-C, o entre Apo B o Lp(a) con LDL-C al tiempo que ambos subtipos de apolipoproteínas se vincularon entre sí de manera negativa. La única correlación positiva significativa para el nivel de triglicéridos se encontró con la concentración de Apo B. Los resultados fueron congruentes con el rol que desempeñan estos marcadores lipídicos (y las apolipoproteínas asociadas) en los procesos metabólicos de transporte de colesterol hacia la periferia y viceversa: transporte reverso periferia Û hígado Û ruta excretoria.
Considerando ahora la variable que resultó de mayor interés –MMSE– se llevó a cabo en primer término una correlación con los biomarcadores del perfil de apolipoproteínas (Tabla XVII) y luego un estudio de regresión ajustada y correlación con el resto de las variables del metabolismo del colesterol.
En la Tabla XVII se muestran nuevamente las correlaciones entre apolipoproteínas al solo efecto de otorgar un marco de referencia. Lo más importante es destacar el hallazgo de correlaciones positivas significativas entre MMSE y Apo D o Apo A, que a su vez correlacionaron positivamente entre sí y se hallaron directamente vinculadas al nivel de HDL-C tal como se mostró en los resultados anteriores. Por otro lado, se observó una correlación negativa entre MMSE y Apo B o Lp(a) (que correlacionó positivamente entre sí y estuvo íntimamente asociada al nivel circulante de LDL-C e indirectamente vinculada con la concentración de HDL-C). Teniendo en cuenta los resultados de estos últimos análisis se procedió a regresionar y correlacionar los datos de MMSE con los marcadores del metabolismo del colesterol que usualmente se emplean en la clínica. En primer término se examinó la variable colesterol total (CT) (Fig. 2).
Tabla XVII. Correlación entre pares de variables seleccionadas (preajustadas por tiempo de evolución)para pacientes en relación al valor del MMSE y lipoproteínas


Figura 2. Regresión y correlación entre MMSE y CT (preajustadas por tiempo de evolución) para el grupo EA
En forma de línea de guiones se marcó la zona de corte (ZC) promedio para MMSE y en línea de puntos el valor máximo de esa variable.
Por debajo de la zona de corte (solo hay pocos pacientes por encima o sobre la misma línea ZC que correspondieron a DCL) se observó una nube de puntos con un débil patrón de correlación entre MMSE y CT que sugirió la inutilidad de CT como un predictor de los valores de MMSE obtenibles. Por ejemplo, considerando para CT el valor límite de 200 mg/dL el MMSE cambió en un rango tan amplio como 10 a 26 (valores que reflejarían desde un leve problema cognitivo hasta una demencia clínicamente muy importante). En la Figura 2 se muestra claramente que el 65% del total de los pacientes (n=122) se encontró dentro del rango de concentraciones de CT comprendido entre 200 y 300 mg/dL sin que esto se asocie indudablemente a valores acotados de MMSE. Solo el 16% mostró valores inferiores a 200 mgdL y el 19% tuvo valores muy patológicos por encima de 300 mg/dL. Los ensayos de correlaciones lineales o no lineales no arrojaron datos significativos. Se prosiguió entonces a investigar la correlación entre MMSE y las sub-fracciones del transporte del colesterol plasmático previo ajuste de cada variable por el tiempo registrado de evolución desde el diagnóstico hasta la toma de muestra (con el error o la limitación derivada del hecho que este tiempo nunca es del todo confiable debido a la dilación que existe entre la percepción de la queja cognitiva –a veces muy subjetiva– y el diagnóstico neurológico propiamente dicho). No obstante las limitaciones que esto pudiera tener, se obtuvo una clara correlación positiva y significativa (p<0,02) entre MMSE y HDL-C (Fig. 3).
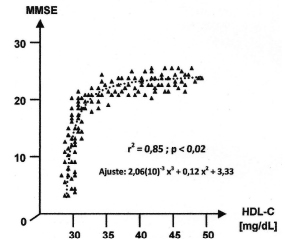
Figura 3. Regresión y correlación entre MMSE y HDL-C (preajustadas por tiempo de evolución) para el grupo EA
A pesar de la claridad matemática de la correlación, ésta no fue en absoluto lineal sino que se ajustó a una curva polinomial de tercer grado. Esto sugirió que a valores muy bajos o bajos de HDL-C existió un gran efecto sobre la predicción (correlación casi lineal) de MMSE, pero a valores superiores a 35 la variable para la cognición entró en una meseta casi insensible a la variación de la concentración del colesterol transportado por HDL. Similarmente se encontró una correlación fuerte con la fracción LDL-C. Si se observa su conducta (Fig. 4), destaca su comportamiento casi especular respecto del obtenido para su contraparte metabólica el HDL-C. En este caso el ajuste se produjo también a una ecuación cuadrática pero se trató de una inversa polinomial de tercer orden.
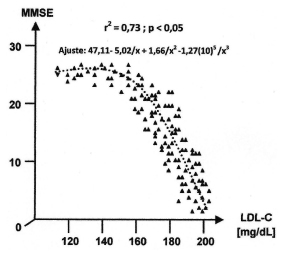
Figura 4. Regresión y correlación entre MMSE y LDL-C (preajustadas por tiempo de evolución) para el grupo EA
Tal como se indicó para el caso del HDL-C el efecto de la variable LDL-C sobre MMSE es disímil según sea el rango de concentraciones considerado. Hay poco efecto deletéreo cuando los valores de LDL-C son bajos o muy bajos y luego de superar aproximadamente los 170 mg/dL se produjo una pronunciada influencia negativa (asintóticamente lineal) sobre los valores de MMSE. Una variable que vincula al metabolismo lipídico con el daño pro-oxidativo (característico en pacientes EA) es la concentración plasmática de LDL oxidado (Ox-LDL). Esta no se ha constituido aún en una determinación clínica rutinaria; sin embargo, amerita un análisis especial porque su vinculación con el desempeño cognitivo estimado por medio del MMSE es más potente aún que todas las variables analizadas hasta ahora (Fig. 5).

Figura 5. Regresión y correlación entre MMSE y Ox-LDL (preajustadas por tiempo de evolución) para el grupo EA
Quizás esta evidencia resulte útil para apoyar su introducción en la práctica clínica como un parámetro confiable de tipo predictor o como control evolutivo de los pacientes con EA. Las otras variables críticas son las referidas al nivel de cobre total (Cu) y de cobre libre no ligado a ceruloplasmina (NCBC). La correlación entre MMSE y Cu proporcionó una nube de puntos con una respuesta poco clara y de bajo nivel de correlación (r2=0,35 para una significación p<0,07; no mostrado). Esta conducta fue algo similar a lo acontecido para el caso de CT pero aún menos definida en términos de tendencia. Sin embargo, no sucedió lo mismo cuando se consideró la concentración de NCBC que mostró la significativa correlación lineal negativa (con MMSE) que se muestra en la Figura 6. El NCBC es otra variable que no integra los protocolos de análisis de pacientes neurológicos y que debería ser tenida en cuenta atendiendo a su clarísima correlación lineal.

Figura 6. Regresión y correlación entre MMSE y NCBC (preajustadas por tiempo de evolución) para el grupo EA
Los valores determinados para MMSE se estudiaron en función de su posible correlación con los datos del cociente bA(1-42)/(1-40), para lo cual fueron ponderados como ya se mencionó, ajustados al mejor polinomio, y evaluados en términos de significancia estadística (Fig. 7). Se observó una significativa correlación inversa que mostró una zona inicial de aparente insensibilidad entre las variables (rango de valores del cociente MMSE que no cambiaron sustancialmente con los valores de bA) y que se localizó dentro de los primeros estadios de la enfermedad (DCL/L-EA). Sin embargo, de forma abrupta se constató una fuerte caída casi lineal hacia valores compatibles con cuadros más avanzados de EA. El perfil de esta curva de ajuste fue sugestivamente similar al hallado para la correlación entre MMSE y niveles plasmáticos de LDL-C y respondió al mismo tipo de polinomio (inverso de tercer orden). Nótese especialmente que la zona de rápido declive se inició en valores del cociente bA que fueron cercanos a 17 (coincidentes con los valores de la Tabla XIV en donde este parámetro se hizo discriminativo para pacientes con EA intermedio y severo).

Figura 7. Regresión y correlación entre MMSE y bA(1-42)/(1-40 (preajustadas por tiempo de evolución) para el grupo EA
Finalmente, se estudió la influencia conjunta de las variables lineales estadísticamente más potentes (Ox- LDL y NCBC) sobre MMSE. Para eso se definió la función auxiliar FAE (función de adición de efectos) definida como la suma de las variables Ox-LDL y NCBC normalizadas en términos de unidades de desviación estándar (Pe = [P-Ppg] / DEpg); FAE = Pe (Ox-LDL) + Pe (NCBC). La atribución de definir una adición (y no otra operación aritmética) descansó en la respuesta lineal confiable de ambas variables continuas frente al MMSE. Por otro lado, la normalización empleada contempló que ambas variables ponderen de manera semejante en el resultado final de la adición pues Ox- LDL cambió entre los extremos 3,0 y 11,5 mientras que NCBC varió dentro del rango 3,4 y 15,0. De modo que fue esperable que FAE tomara algún valor (en el espacio continuo) entre 6 y 27. De esta forma se obtuvo el análisis que se muestra en la Figura 8.

Figura 8. Regresión y correlación entre MMSE y FAE para el grupo EA
En este análisis (a pesar de ser obtenido a partir de una variable de cálculo o impura) claramente se observó un refuerzo de la correlación lineal negativa. La dependencia entre FAE y MMSE superó a las halladas para las variables Ox-LDL y NCBC por separado. La fuerte correlación de alta significancia sugirió que ambos factores ejercen un efecto aditivo (no lo podríamos catalogar estrictamente de sinérgico debido a que se mantiene y consolida el ajuste lineal) sobre el decrecimiento que el Ox-LDL y el NCBC conjuntamente fueron capaces de ejercer sobre la variable MMSE. Así, la determinación combinada (normalizada y aditiva) de Ox-LDL y de NCBC se constituyó sin duda alguna en el mejor marcador clínico para el deterioro cognitivo de entre todos los explorados en este estudio.
Discusión y Conclusiones
Es bien sabido que realizar protocolos experimentales en humanos resulta sumamente complejo debido a la cantidad de variables que se escapan a un control estricto e introducen dispersión en los resultados. En el presente estudio se siguió un protocolo de inclusión/exclusión sumamente exigente y en consecuencia se desestimó la inclusión de un número importante de controles y pacientes con el fin de minimizar este problema y otorgar mayor solidez bioestadística a los resultados. Para esto se siguieron recomendaciones internacionales (113)(148). En la parte de resultados experimentales ya se ha descripto y caracterizado a la población estudiada, por lo que a continuación se hará hincapié en la cuestión central del trabajo. Según lo mencionado en la introducción, la hipótesis de trabajo descansó en suponer que existe una asociación o correlación estadísticamente demostrable entre la pérdida del desempeño cognitivo (atribuible a daño neuronal/sináptico) y los niveles circulantes de cobre y colesterol. Estos dos factores podrían causar un efecto aditivo o potenciador de la pérdida neuronal y deberían reflejarse en cambios demostrables en la evaluación cognitiva mediante MMSE y/o en los valores de biomarcadores periféricos asociados al proceso amiloidogénico.
Los factores de origen genético que predisponen a la EA, o que contribuyen a su etiopatogenia y luego a la progresión del deterioro cognitivo, son complejos de determinar y en general costosos y difíciles de implementar como mecanismos de detección temprana y masiva en periodos asintomáticos y en grandes poblaciones (149)(150). De ahí que estudios tendientes a identificar biomarcadores pronósticos que sean accesibles y confiables se hayan vuelto una prioridad a nivel internacional, especialmente en el tercer mundo. Los factores epigenéticos como los estudiados aquí (colesterol transportado por diferentes apolipoproteínas, marcadores de estrés oxidativo y cobre libre en sangre periférica) son determinaciones cada vez más sencillas de implementar en el laboratorio de Bioquímica Clínica de mediana complejidad y por lo que ya se ha visto, tienen un gran potencial en la práctica asistencial (117)(151-158). En este estudio se ha demostrado que la homeostasis del Cu se encuentra seriamente alterada en los pacientes con EA y que esto resulta en la elevación de los niveles de Cu total y Cu libre respecto de los hallados en la población general sana. Los resultados indican que la fracción de Cu circulante no complejada a ceruloplasmina sería la responsable directa de los daños observados en la neurodegeneración tal como ha sido sugerido por varios estudios previos efectuados en animales y humanos (16) (18)(43-47)(78)(117)(160)(161). Probablemente este efecto se extienda a otras proteínas captadoras de Cu como las metalotioneínas (MTs), y varios polipéptidos del muy complejo sistema homeostático que posee este metal en el organismo (162-176). Varias investigaciones demostraron que el Cu se libera durante el proceso normal de neurotransmisión y alcanza en el espacio intersináptico concentraciones zonales de hasta 15 μM (112)(168)(177)(178) (lo que resulta varias veces superior a lo hallado en otras zonas histológicas, o en zonas no sinápticas del tejido cerebral) y también desde hace tiempo existe evidencia que demuestra que este metal favorece la deposición de los agregados de péptidos amiloides (bA) tanto in vitro como in vivo (15)(17)(56) (162)(175)(179-181). A su vez, la acumulación amiloidogénica causa estrés oxidativo en el SNC (12)(15)(60) (62)(63)(101)(182)(183). Estos efectos perjudiciales se ven potenciados por varias razones específicas que provocan la acumulación de especies radicalarias (ROS y RNS, especialmente). Dentro de la lista de razones se destacan una tasa metabólica muy elevada, altos niveles de metales de transición capaces de catalizar la formación del radical hidroxilo por medio de reacciones tipo Fenton, preponderancia de ácidos grasos poliinsaturados (PUFAs), presencia de neurotransmisores auto-oxidables, y sobre todo un sistema de defensa antioxidante muy poco efectivo (38). En relación con esta cuestión, existe un debate acerca de si es el entorno pro-oxidativo el que favorece la deposición amiloidogénica, o por el contrario, la formación de placas de bA son las que promueven la oxidación de metabolitos con formación de TBARs, PCs, etc. (184). A pesar de que esta cuestión lleva años en discusión, aún no hay evidencia contundente para ratificar una u otra postura y lo más factible es que ambas sean ciertas (101)(183-185).
La dishomeostasis del cobre también está involucrada en la etiopatogénesis de otras demencias como las vasculares y enfermedades neurodegenerativas que afectan tanto al SNC como al SNP (64)(65)(117)(161) (162(184-186). Por otro lado, el colesterol también ha sido vinculado a más de un tipo de enfermedad neurodegenerativa incluyendo la EA (56)(59)(66)(72)(74) (75)(99)(100)(187). Es bien conocido que el colesterol juega un rol central en el desarrollo normal y el mantenimiento de la fisiología del SNC (189-192). Baste mencionar que el cerebro representa sólo el 2% del peso corporal de un adulto normal mientras que allí se acumula el 25% del total de colesterol presente en todo el organismo (193). El papel del colesterol en la etiopatogénesis de la EA llevó a diseñar varias estrategias para poder controlar o disminuir la incidencia/prevalencia de esta enfermedad, empleándose desde medidas dietéticas (6) (11) (54) (70) (100) (103) (106) (194-196), hasta la prescripción de estatinas (59)(69)(106), ambas sin un éxito fehaciente (70). Estos intentos descansaron en evidencia que sugería que aún muy leves incrementos del colesterol en sangre periférica causaban un aumento significativo de la propensión a desarrollar EA (10). Existe aún controversia sobre el proceso que vincula al colesterol con la formación de péptidos amiloides, aunque desde hace mucho tiempo está claro que hay una interacción directa del metal con el bA por formación de puentes entrecruzados entre restos de tirosinas de la cadena peptídica y oligomerización covalente con drástica disminución de la solubilidad de esos agregados (11)(54)(56)(66-68)(179)(197)(198). Esto último puede tener una decisiva importancia a la hora de analizar el proceso etiológico subyacente en las neurodegeneraciones, en especial en la acontecida en la EA, ya que de ser cierta la hipótesis de un sistema homeostático bA-dependiente, pequeñas elevaciones del NCBC podrían causar significativas acumulaciones de material amiloideo. Al menos en animales esto es cierto, ya que cantidades traza de Cu inducen la acumulación de péptido bA y la formación de placas seniles en cerebro de conejos hipercolesterolémicos (49- 52) seguida de la inducción por parte del bA de daño pro-oxidativo y pro-apoptótico. Más interesante aún es el hecho que reciente información experimental sostiene con evidencia bioquímica y biofísica de un modelo de posible sinergia cobre-colesterol, al menos en ratas de la cepa Wistar alimentadas con dietas enriquecidas en cobre y en colesterol (14)(20)(65)(199). También en modelos animales este metal produce grandes alteraciones en el sistema de defensa antioxidante, tanto enzimático como no enzimático, y provoca incremento de todos los marcadores de estrés oxidativo/nitrativo (peroxidación lipídica y carbonilación de proteínas, entre otros) lo cual también se observó en este estudio. Este escenario activa señales pro-apoptóticas mediadas por caspasa-3 y otras rutas de muerte celular programada dependientes de mili- y micro-calpaínas, al tiempo que se incrementa la muerte por necrosis y ocurren severos daños de la calidad de los acilos grasos unidos a lípidos complejos con disminución de ácidos grasos polienoicos y enriquecimiento de los saturados (20)(65) (161)(196). Muchos de estos biomarcadores de estrés oxidativo/nitrativo (tal como los definen Mangialasche et al. (152) que se alteran en estos sistemas experimentales también fueron analizados en muestras de plasmas de sangre periférica de pacientes con diversos cuadros neurodegenerativos como el mal de Alzheimer, mal de Parkinson, y en demencias de origen vascular (20)(44) (78)(117)(161).
Para darle sentido al posible papel del Cu en estos procesos se investigó también el impacto de la polución por este metal en muestras de plasma de personas pertenecientes a poblaciones profesionalmente expuestas (20)(130)(199) y en mujeres usuarias de dispositivos intrauterinos a base de Cu (Cu-DIU) (20)(200)(201) encontrándose sorprendentes incrementos del metal en sangre periférica conjuntamente con varias alteraciones del sistema redox (antioxidantes y enzimas de defensa antioxidante, proteínas transportadoras de cobre en sangre periférica, etc.). En relación a esta cuestión sería muy interesante llevar a cabo un estudio retrospectivo a gran escala de la incidencia y prevalencia de los diferentes tipos de demencias entre estas poblaciones expuestas a sobrecarga de Cu en comparación con grupos de referencia no expuestos. Hasta el presente y en forma sucinta las principales conclusiones a las que se arribó en los estudios con humanos fueron las siguientes: (1) una estadísticamente significativa asociación entre el Cu plasmático y cuadros neurodegenerativos, en especial EA; (2) elevados niveles de Cu en parientes directos de pacientes con demencias vasculares y correlación de este biomarcador en la progresión del cuadro clínico; (3) elevación del nivel plasmático de la proteína transportadora de Cu ceruloplasmina (CRP) en todos los grupos de pacientes dementes en respuesta al componente inflamatorio de la neurodegeneración; (4) progresivo incremento de la concentración de otras proteínas involucradas en la homeostasis del Cu (en especial MTs) en relación con la gravedad del cuadro demencial; (5) incrementos de los cocientes Cu/CRP y Cu/MTs como indicadores de posible utilidad clínica en la progresión de EA, pero no así en el mal de Parkinson o en las demencias vasculares (biomarcadores diferenciales); (6) correlación entre la prueba de desempeño cognitivo MMSE y los niveles de Cu libre (NCBC) con dependencia lineal para EA y mal de Parkinson, y polinomial para demencias vasculares; (7) clara dependencia del progreso de EA con el marcador NCBC; y (8) valor predictivo de la determinación de Cu y del cociente Cu/CRP como evaluador de riesgo en parientes directos de pacientes. Esta evidencia surgió a partir de estudios anteriores y de la presente investigación (20)(45-46)(78)(102)(117)(161), de modo que se ajusta y concuerda con los resultados aquí presentados específicamente para el caso de la EA y prometen ser de utilidad en la transferencia a la práctica asistencial.
En cuanto al mecanismo íntimo del daño que causa la asociación hipercolesterolemia – hiper NCBC (o elevación de la función de adición FAE) es posible decir que se ha comenzado a vislumbrar cuál sería su ruta de progresión a partir de resultados en experimentación animal y en cultivos celulares (14)(20)(65)(66) (82)(199). El fosfolípido acídico cardiolipina (CL) es muy abundante en membranas mitocondriales de cerebro de ratas Wistar alimentadas con dietas enriquecidas en Cu y colesterol. Este fosfolípido mostró que sus cadenas laterales de ácidos grasos se encuentran más insaturadas en el SNC (enriquecidas en PUFAs sobre todo 22:5 y 22:6 de la serie esencial n-3). La mayor peroxidabilidad de la CL inducida por NCBC se encontró al analizar la corteza, el hipocampo, y la sustancia nigra (20)(65)(66). De modo que la CL peroxidada podría ser el agente causativo directo del daño neurológico al funcionar como señal precursora de la integración del apoptosoma y de la pérdida neuronal por muerte programada vía liberación de citocromo c desde la mitocondria hacia el citoplasma (82)(202-205). Esto es altamente factible a la luz de recientes evidencias biofísicas presentadas por Arnal et al (65)(66) que demostraron (en este modelo animal de sobrecarga de Cu y colesterol) una alteración de la permeabilidad y de la estructura de las membranas mitocondriales mediante determinaciones de microviscosidad aparente y potenciales trans-membrana (∆Ψ). La asociación de altos niveles de NCBC y colesterol provocaría la peroxidación de CL de un modo potenciado y, finalmente, la activación funcional del apoptosoma con la subsecuente pérdida sináptica y de cuerpos neuronales. Ese modo de acción está directamente vinculado no solo al colesterol per se sino también a la formación de productos de peroxidación del colesterol dentro del SNC, tales como por ejemplo los oxicolesteroles y los secosteroles aldehídicos que se sabe contribuyen a conformar el apoptosoma y dañar la barrera hematoencefálica (73)(74)(103) (105)(213). Todos los fenómenos pro-oxidativos del sistema nervioso central terminan desencadenando un cuadro inflamatorio crónico (gliosis reaccional) que se sabe concluye en muerte celular programada (31)(32) (65)(66)(80)(82)(87)(108)(109)(111)(117)(208-211). Algo similar acontece con la etiopatogénesis de otras enfermedades neurodegenerativas (89)(90)(212). Una cuestión crucial que merece ser estudiada a la luz de este mecanismo es el papel desempeñado por la barrera hematoencefálica. Se conoce que el colesterol periférico está mayormente esterificado y que, en cambio, el cerebral está en enorme proporción al estado libre (65)(66)(71)(72)(206)(207). También se sabe que la barrera hematoencefálica funciona regulando la homeostasis del cobre de un modo altamente sofisticado (175)(176)(208). De modo que la alteración de ambos factores epigenéticos en la sangre periférica deben producir algún tipo de daño (muy probablemente de causa pro-oxidativa) en la integridad y en la función de la barrera hematoencefálica para permitir que esos cambios orquesten las alteraciones antes descriptas dentro del SNC. Ya existen algunas evidencias en animales que soportan esta suposición y que podrían también darse en el caso de humanos (14)(20)(66)(169), aunque es un campo de investigación que sin dudas requiere de muchos más esfuerzos para ser esclarecido. Esta teoría del daño pro-oxidativo específicamente sobre la estructura –y función– de la barrera hematoencefálica causado por agentes pro-oxidantes como el NCBC (45)(213) (214) y los oxicolesteroles (73)(74)(103)(105)(213) ha sido la base sobre la cual se implementaron en la práctica clínica terapias antioxidantes en pacientes con EA (17)(83)(113)(215-219).
Finalmente, resulta pertinente hacer un comentario sobre la prevalencia e incidencia de la hipercolesterolemia y de la hipercupremia en este medio y en el mundo para darle mayor pertinencia clínica a los resultados de este estudio. El requerimiento de Cu en humanos es de aproximadamente 1,0 mg/día (depende del IMC, del sexo, la raza, la edad y el estado clínico general), y la RDA (Recommended dietary allowances) establecida por la FDA (Food and Drug Administration, EE.UU.) tiene como valores límites máximos aceptables 1,80 mg/día (20) (65)(66). En el Reino Unido, Australia y Europa la RDA desciende a valores de 0,95 mg/día. La dieta occidental típica –sobre todo en América Latina y el Caribe– puede llegar a contener hasta 4,20 mg cada 2.000 calorías/ día (20). La OMS (Organización Mundial de la Salud) estimó las necesidades de ingesta mínima diaria para el cobre en humanos en 0,60 mg/día para mujeres y 0,70 mg/día para hombres y sugiere no sobrepasarlas (220). Lo concreto es que en la actualidad aún no se cuenta con datos certeros sobre los índices LOAEL (Lowest Observed Adverse Effect Level) o NOAEL (Non-Observed Adverse Effect Level) que hayan sido determinados de manera confiable mediante experimentación en humanos. En relación con este tema, baste mencionar que suplementos minerales y vitamínicos de venta libre en Argentina y varios países del mundo contienen hasta 3 mg de Cu inorgánico por dosis y suelen recomendarse dos o más dosis diarias (20)(66). Como ya se dijo, el uso del DIU es otra fuente común de exposición a Cu inorgánico (20) (200)(201) (especialmente oxicloruro de cobre) y quizás en esa práctica anticonceptiva tan difundida se asiente la explicación de la prevalencia de EA en mujeres (222) sobre todo si han sido hipercolesterolémicas. Se sabe desde hace años que el uso del DIU conlleva la liberación de Cu inorgánico (o libre) a la circulación sanguínea comprendida dentro del rango 43-83 μg Cu/día, y esta cantidad podría ser aún mayor dependiendo del tipo de dispositivo de cobre usado (223)(224). Algunos investigadores han propuesto que pequeños incrementos de Cu plasmático de solo 1 μmol/L correlacionan con un 80% de aumento de riesgo para contraer la EA y una significativa disminución de las funciones neurofisiológicas centinelas y de desempeño cognitivo (47) (225). A esta cuestión de la sobrecarga involuntaria de Cu no ligado, se le debe sumar la epidemia de hipercolesterolemia que viene sufriendo la humanidad desde hace décadas y para lo cual no es necesario abundar en datos epidemiológicos (226)(227). La investigación de las fuentes posibles de sobrecarga de Cu en humanos dejó en claro que existen tres que son relevantes: (i) suplementación mineral de venta libre, (ii) polución ambiental (incluyendo aquí actividades laborales, residencia en cercanías a ciertas manufacturas como las de empresas de electrónicos y esmaltados, empleos de dispositivos de amalgamas de cobre en el organismo, etc.), y (iii) fundamentalmente el agua de bebida y de cocción de alimentos (20). En todo el mundo y desde hace años se insiste en la investigación y en un control mucho más exhaustivo del nivel de Cu en aguas de uso humano/domiciliario (40)(49)(50)(82)(88)(91)(92) (95)(96)(187)(221), sin que toda esta evidencia experimental y epidemiológica haya tenido hasta ahora un impacto decisivo en países que no pertenezcan al primer mundo (20)(91). En la ciudad Argentina de La Plata y sus alrededores se encontraron extensas zonas con elevados niveles de Cu inorgánico en forma de varios óxidos y sales presentes en el agua de bebida y de cocción. Esta es una cuestión hasta ahora postergada de la Medicina preventina que requiere urgente intervención para su remedio.
Se concluye que la hipótesis planteada en este estudio resultó ser estadísticamente cierta, es decir: existe una correlación significativa y directamente proporcional entre el declive de la función cognitiva en pacientes con EA, la alteración del metabolismo de las proteínas amiloides y los niveles de Cu no complejado y colesterol (en especial el colesterol transportado por Ox-LDL). Es posible combinar estas dos últimas variables en una función de adición de efectos (FAE) que promete ser una herramienta extrapolable a la Medicina asistencial. Debe recordarse que la muerte neuronal por apoptosis y la pérdida de sinaptosomas son procesos silentes, asintomáticos y asignológicos hasta cierta etapa en la cual los recursos curativos no existen y solo se puede echar mano de estrategias paliativas. No obstante, estos acontecimientos muchas veces se pueden ver reflejados en la sangre periférica tal como lo demuestra el presente estudio y otros (20)(43)(44)(65)(66)(118) (161). De allí que sea altamente pertinente insistir con la investigación y la validación de biomarcadores que sean útiles en la práctica clínica por poseer capacidad diagnóstica o pronóstica. Este punto de vista coincide con el de Pal (91), Schitti (49), Arnal (66), Brewer (26) y muchos otros investigadores que señalan insistentemente la urgencia de contar con nuevos marcadores de laboratorio que aborden el problema de riesgo que significa la exposición crónica y simultánea a Cu libre e hipercolesterolemia. Al día de hoy es cada vez mayor la acumulación de evidencia experimental que vincula a estos dos factores epigenéticos con la etiopatogenia de la EA. Obviamente, esto ayudaría a sustentar mejores políticas de saneamiento ambiental y campañas preventivas dentro de poblaciones en riesgo. Futuros estudios en grandes muestras ayudarán a esclarecer el valor de los hallazgos aquí presentados.
AGRADECIMIENTO
Al CONICET y a la UNLP por los subsidios oportunamente concedidos a los Prof. Dres. Lausada y Marra. Al Prof. Dr. Antonio Sánchez Sandoval (Profesor de Bioestadística de la Universidad Autónoma de México), por haber revisado el procesamiento de los resultados experimentales.
1. St. John PD, Montgomery PR, Kristjansson B, McDowell L. Cognitive scores, even within the normal range, predict death and institutionalization. Age Ageing 2002; 31: 373-8. [ Links ]
2. Ewbank DC. Deaths attributable to Alzheimer’s disease in the United States. Am J Public Health 1999; 89 (1): 90-2.
3. Mangone CA. Heterogeneidad clínica en la enfermedad de Alzheimer. Diferentes perfiles clínicos pueden predecir el intervalo de progresión Rev Neurol 2004; 42 (38): 675-81. [ Links ]
4. Mangone CA, Genovese OM, Abel CG. Trastornos cognitivo conductuales en el período crónico de la intoxicación por neurotóxicos laborales y ambientales. Vertex 2006; Vol XVII: 16-22. [ Links ]
5. Mangone CA, Caravello OL, Bavio JP, Gonzalez MI, Pallo V, Genovese O, et al, editores. Testing the new pathogenic hypothesis of the sporadic form of Alzheimer disease (SAD) in Cognitive Disorders. New York (NY): Frank Colombus Publishers; 2008. Capítulo 10, p. 210-89. [ Links ]
6. Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health 1998; 88 (9): 1337-42.
7. Mansoor MA, Bergmark C, Haswell SJ, Savage IF, Evans PH, Berge RK, et al. Correlation between plasma total homocysteine and copper in patients with peripheral vascular disease. Clin Chem 2000; 46 (3): 385-91. [ Links ]
8. Campbell A. Beta-amyloid: friend or foe. Med Hypotheses 2001; 56 (3): 388-91. [ Links ]
9. Benítez del Rosario MA. Diagnóstico del deterioro cognitivo: dificultades metodológicas en la frontera de la normalidad cognitiva. Atención Primaria 2002; 30 (1): 5-15. [ Links ]
10. Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra Garcia M, Manjon M, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 2003; 61 (2): 199-205. [ Links ]
11. Popp J, Lewczuk P, Kölsch H, Meichsner S, Maier W, Kornhuber J, et al. Cholesterol metabolism is associated with soluble amyloid precursor protein production in Alzheimer’s disease. J Neurochem 2012; 123 (2): 310-6.
12. Porcellotti S, Fanelli F, Fracassi A, Sepe S, Cecconi F, Bernardi C, et al. Oxidative stress during the progression of-amyloid pathology in the neocortex of the Tg2576 mouse model of Alzheimer’s disease. Oxid Med Cell Longev 2015; 967203.
13. Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med 2010; 362 (4): 329-44.
14. Arnal N, Dominici L, de Tacconi MJ, Marra CA. Copper-induced alterations in rat brain depends on route of overload and basal copper levels. Nutrition 2014; 30 (1): 96-106. [ Links ]
15. Bush AI, Masters CL, Tanzi RE. Copper, beta-amyloid, and Alzheimer’s disease: tapping a sensitive connection. Proc Natl Acad Sci U S A. 2003; 100 (20): 11193-4.
16. Bush AI. Copper, zinc, and the metallobiology of Alzheimer disease. Alzheimer Dis Assoc Disord 2003; 17 (3): 147-50. [ Links ]
17. Bush AL, Tanzi RE. Therapeutics for Alzheimer’s disease based on the. Metal hypothesis. J Am Soc Exp NeuroTherapeutics 2008; 5: 421-32.
18. Bush AI. The metal theory of Alzheimer’s disease. J Alzheimer’s Dis 2013; 33 Suppl 1: S277-81.
19. Loeffler DA, LeWitt PA, Juneau PL, Sima AA, Nguyen HU, DeMaggio AJ, et al. Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders. Brain Res 1996; 738 (2): 265-74. [ Links ]
20. Arnal N, Tacconi de Alaniz MJ, Marra CA. Involvement of copper overload in human diseases. En "Metals in Biological Systems". Giménez MS, editora. Kerala, India: Research Signpost, Transworld Research Network; 2010. Capítulo 1, p. 1-28. [ Links ]
21. Ayton S, Lei P, Bush AI. Metallostasis in Alzheimer’s disease. Free Radic Biol Med 2013; 62: 76-89.
22. Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol 2015; 14 (1): 103-13.
23. Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997; 272: 20313-6. [ Links ]
24. Praticò D, Delanty N. Oxidative injury in diseases of the central nervous system: focus on Alzheimer’s disease. Am J Med 2000; 109 (7): 577-85.
25. Ozcelik D, Uzun H. Copper intoxication; antioxidant defenses and oxidative damage in rat brain. Biol Trace Elem Res 2009; 127 (1): 45-52. [ Links ]
26. Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. Biofactors 2012; 38 (2): 107-13.
27. Choo XY, Alukaidey L, White AR, Grubman A. Neuroinflammation and copper in Alzheimer’s disease. Int J Alzheimer’s Dis 2013: 145345.
28. Hordyjewska A, Popiołek Ł, Kocot J. The many "faces" of copper in medicine and treatment. Biometals 2014; 27 (4): 611-21. [ Links ]
29. Dubinina EE, Schedrina LV, Neznanov NG, Zalutskaya NM, Zakharchenko DV. Oxidative stress and its effect on cells functional activity of Alzheimer’s disease. Biomed Khim 2015; 61 (1): 57-69.
30. Kanamaru T, Kamimura N, Yokota T, Iuchi K, Nishimaki K, Takami S, et al. Oxidative stress accelerates amyloid deposition and memory impairment in a double-transgenic mouse model of Alzheimer’s disease. Neurosci Lett 2015; 587: 126-31.
31. Moneim AE. Oxidant/Antioxidant imbalance and the risk of Alzheimer’s disease. Curr Alzheimer Res 2015; 12 (4): 335-49.
32. Matsumura A, Emoto MC, Suzuki S, Iwahara N, Hisahara S, Kawamata J, et al. Evaluation of oxidative stress in the brain of a transgenic mouse model of Alzheimer’s disease by in vivo electron paramagnetic resonance imaging. Free Radic Biol Med 2015; 85: 165-73.
33. Markesbery WR, Carney JM. Oxidative alterations in Alzheimer’s disease. Brain Pathol 1999; 9 (1): 133-46.
34. Cerpa W, Varela-Nallar L, Reyes AE, Minniti AN, Inestrosa NC. Is there a role for copper in neurodegenerative diseases? Mol Aspects Med 2005; 26 (4-5): 405-20. [ Links ]
35. Collins JF, Klevay LM. Copper. Adv Nutr 2011; 2 (6): 520-2. [ Links ]
36. Agostinho P, Pliássova A, Oliveira CR, Cunha RA. Localization and trafficking of amyloid-βprotein precursor and secretases: Impact on Alzheimer’s disease. J Alzheimer’s Dis 2015; 45 (2): 329-47.
37. Ahuja A, Dev K, Tanwar RS, Selwal KK, Tyagi PK. Copper mediated neurological disorder: visions into amyotrophic lateral sclerosis, Alzheimer and Menkes disease. J Trace Elem Med Biol 2015; 29: 11-23. [ Links ]
38. Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Free Radicals in Biology and Medicine. Fifth Edition, Halliwell B y Gutteridge JMC, editores. Tercera Edición, Oxford Science Publications. ISBN: 9780198717485, 2015. [ Links ]
39. Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci 2001; 21 (12): 4183-7. [ Links ]
40. Sayre LM, Perry G, Atwood CS, Smith MA. The role of metals in neurodegenerative diseases. Cell Mol Biol (Noisy-le-grand) 2000; 46 (4): 731-41. [ Links ]
41. Becaria A, Lahiri DK, Bondy SC, Chen D, Hamadeh A, Li H, et al. Aluminum and copper in drinking water enhance inflammatory or oxidative events specifically in the brain. J Neuroimmunol 2006; 176 (1-2): 16-23. [ Links ]
42. Aliev G, Priyadarshini M, Reddy VP, Grieg NH, Kaminsky Y, Cacabelos R, et al. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer’s disease. Curr Med Chem 2014; 21 (19): 2208-17.
43. Squitti R, Barbati G, Rossi L, Ventriglia M, Dal Forno G, Cesaretti S, et al. Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF [beta]-amyloid, and h-tau. Neurology 2006; 67 (1): 76-82. [ Links ]
44. Squitti R. Metals in Alzheimer’s disease: a systemic perspective. Front Biosci, Landmark MG, editor. 17: 451-72; 2012.
45. Squitti R, Hoogenraad T, Brewer G, Bush AI, Polimanti R. Copper status in Alzheimer’s disease and other neurodegenerative disorders. Int J Alzheimer’s Dis 2013: 838274.
46. Squitti R, Ghidoni R, Siotto M, Ventriglia M, Benussi L, Paterlini A, et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann Neurol 2014; 75 (4): 574-80. [ Links ]
47. Squitti R, Simonelli I, Ventriglia M, Siotto M, Pasqualetti P, Rembach A, et al. Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J Alzheimer’s Dis 2014; 38 (4): 809-22.
48. Squitti R, Siotto M, Polimanti R. Low-copper diet as a preventive strategy for Alzheimer’s disease. Neurobiol Aging 2014; 35 Suppl 2: S40-50.
49. Squitti R. Copper subtype of Alzheimer’s disease (AD): meta-analyses, genetic studies and predictive value of non-ceruloplasmim copper in mild cognitive impairment conversion to full AD. J. Trace Elem Med Biol 2014; 28 (4): 482-5.
50. Sparks DL, Schreurs BG. Trace amounts of copper in water induce beta-amyloid plaques and learning deficits in a rabbit model of Alzheimer’s disease. Proc Natl Acad Sci USA 2013; 100 (19): 11065-9.
51. Sparks DL. The early and ongoing experience with the cholesterol-fed rabbit as a model of Alzheimer’s disease: the old, the new and the pilot. J Alzheimer’s Dis 2008; 15 (4): 641-56.
52. Sparks DL, Ziolkowski C, Lawmaster T, Martin T. Influence of water quality on cholesterol-induced tau pathology: preliminary data. Int J Alzheimer’s Dis 2011: 987023.
53. Gerber H, Wu F, Dimitrov M, Garcia Osuna GM, Fraering PC. Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-b peptide production. J Biol Chem 2017; 292 (9): 3751-67.
54. Rao KSJ, Hegde ML, Anitha S, Musicco M, Zucca FA, Turro NJ, et al. Amyloid b and neuromelanin—Toxic or protective molecules? The cellular context makes the difference. Progr Neurobiol 2006; 78: 364–73.
55. Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis 2008; 29 (3): 422-37.
56. Hooper NM. Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein. Biochem Soc Trans 2005; 33 (Pt 2): 335-8. [ Links ]
57. Beel AJ, Sakakura M, Barrett PJ, Sanders CR. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships?. Biochim Biophys Acta 2010; 1801 (8): 975-82.
58. Walter J, Van Echten-Deckert G. Cross-talk of membrane lipids and Alzheimer-related proteins. Mol Neurodegener 2013; 8: 34. [ Links ]
59. Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 2014; 71 (2): 195-200. [ Links ]
60. Zambón D, Quintana M, Mata P, Alonso R, Benavent J, Cruz-Sánchez F, et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am J Med 2010; 123 (3): 267-74. [ Links ]
61. Cai Z, Zhao B, Ratka A. Oxidative stress and b-amyloid protein in Alzheimer’s disease. Neuromol Med 2011; 13 (4): 223-50.
62. De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer’s disease. Subcell Biochem 2012; 65: 329-52.
63. Giacovazzi R, Ciofini I, Rao L, Amatore C, Adamo C. Copper-amyloid-b complex may catalyze peroxynitrite production in brain: evidence from molecular modeling. Phys Chem Chem Phys 2014; 16 (21): 10169-74. [ Links ]
64. Ill-Raga G, Ramos-Fernández E, Guix FX, Tajes M, Bosch-Morató M, Palomer E, et al. Amyloid-b peptide fibrils induce nitro-oxidative stress in neuronal cells. J Alzheimer’s Dis 2010; 22 (2): 641-52.
65. Arnal N, Castillo O, de Alaniz MJ, Marra CA. Effects of copper and/or cholesterol overload on mitochondrial function in a rat model of incipient neurodegeneration. Int J Alzheimer’s Dis 2013: 645379.
66. Arnal N, Morel GR, de Alaniz MJ, Castillo O, Marra CA. Role of copper and cholesterol association in the neurodegenerative process. Int J Alzheimer’s Dis 2013: 414817.
67. Chauhan NB. Membrane dynamics, cholesterol homeostasis, and Alzheimer’s disease. J Lipid Res 2003; 44 (11): 2019-29.
68. Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012; 336 (6085): 1168-71. [ Links ]
69. Lee HJ, Korshavn KJ, Kochi A, Derrick JS, Lim MH. Cholesterol and metal ions in Alzheimer’s disease. Chem Soc Rev 2014; 43(19): 6672-82.
70. Wanamaker BL, Swiger KJ, Blumenthal RS, Martin SS. Cholesterol, statins, and dementia: what the cardiologist should know. Clin Cardiol 2015; 38 (4): 243-50. [ Links ]
71. Wood WG, Li L, Müller WE, Eckert GP. Cholesterol as a causative factor in Alzheimer’s disease: a debatable hypothesis. J Neurochem 2014; 129 (4): 559-72.
72. Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol 2001; 12 (2): 105-12. [ Links ]
73. Leoni V, Caccia C. Oxysterols as biomarkers in neurodegenerative diseases. Chem Phys Lipids 2011; 164 (6): 515-24. [ Links ]
74. Leoni V, Masterman T, Patel P, Meaney S, Diczfalusy U, Björkhem I. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J Lipid Res 2003; 44 (4): 793-9. [ Links ]
75. Nieva J, Song BD, Rogel JK, Kujawara D, Altobel L 3rd, Izharrudin A, et al. Cholesterol secosterol aldehydes induce amyloidogenesis and dysfunction of wild-type tumor protein p53. Chem Biol 2011; 18 (7): 920-7. [ Links ]
76. Vicario A, Del Sueldo M, Fernández RA, Enders J, Zilberman J, Cerezo GH. Cognition and vascular risk factors: an epidemiological study. Int J Hypertens 2012: 783696. [ Links ]
77. Wong BX, Hung YH, Bush AI, Duce JA. Metals and cholesterol: two sides of the same coin in Alzheimer’s disease pathology. Front Aging Neurosci 2014; 6: 91.
78. Cerpa WF, Barría MI, Chacón MA, Suazo M, González M, Opazo Cet al. The N-terminal copper-binding domain of the amyloid precursor protein protects against Cu2+ neurotoxicity in vivo. FASEB J 2004; 18 (14): 1701-3. [ Links ]
79. Squitti R, Polimanti R. Copper phenotype in Alzheimer’s disease: dissecting the pathway. Am. J Neurodegener Dis 2013; 2 (2): 46-56.
80. Viola KL, Klein WL. Amyloid b oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 2015; 129(2): 183-206.
81. Zraika S, Hull RL, Udayasankar J, Aston-Mourney K, Subramanian SL, Kisilevsky R, et al. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009; 52 (4): 626-35. [ Links ]
82. Jiao XT, Liu XQ, Huang LS, Zhang YJ, Han LS. Role of caspase-3, -8, and -9 in apoptosis of copper induced primary cortical neurons. Zhongguo Dang. Dai Er Ke Za Zhi 2009; 11 (11): 917-22. [ Links ]
83. Grant WB, Campbell A, Itzhaki RF, Savory J. The significance of environmental factors in the etiology of Alzheimer’s disease. J Alzheimer’s Dis 2002; 4 (3): 179-89.
84. Greenough MA, Camakaris J, Bush AI. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 2013; 62 (5): 540-55.
85. Kawahara M, Mizuno D. Crosstalk between metals and neurodegenerative diseases. Nihon. Eiseigaku Zasshi 2014; 69 (3): 155-65. [ Links ]
86. Eife R, Weiss M, Barros V, Sigmund B, Goriup U, Komb D, et al. Chronic poisoning by copper in tap water: I. Copper intoxications with predominantly gastointestinal symptoms. Eur J Med Res 1999; 4 (6): 219-23. [ Links ]
87. Rivera-Mancía S, Pérez-Neri I, Ríos C, Tristán-López L, Rivera-Espinosa L, Montes S. The transition metals copper and iron in neurodegenerative diseases. Chem Biol Interact 2010; 186 (2): 184-9. [ Links ]
88. Eskici G, Axelsen PH. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 2012; 51 (32): 6289-311.
89. Alomary A, Al-Momani IF, Obeidat SM, Massadeh AM. Levels of lead, cadmium, copper, iron, and zinc in deciduous teeth of children living in Irbid, Jordan by ICP-OES: some factors affecting their concentrations. Environ Monit Asses. 2013; 185 (4): 3283-95. [ Links ]
90. Kozlowski H, Kolkowska P, Watly J, Krzywoszynska K, Potocki S. General aspects of metal toxicity. Curr Med Chem 2014; 21 (33): 3721-40. [ Links ]
91. Pal A. Copper toxicity induced hepatocerebral and neurodegenerative diseases: an urgent need for prognostic biomarkers. Neurotoxicology 2014; 40: 97-101. [ Links ]
92. Sullivan MJ, Leavey S. Heavy metals in bottled natural spring water. J Environ Health 2011; 73 (10): 8-13. [ Links ]
93. Pizarro F, Olivares M, Uauy R, Contreras P, Rebelo A, Gidi V. Acute gastrointestinal effects of graded levels of copper in drinking water. Environ Health Perspect 1999; 107 (2): 117-21. [ Links ]
94. Alvarez CCA. Diabetes mellitus y aterosclerosis. Segunda edición, Buenos Aires: EUDEBA, 2006. [ Links ]
95. Eife R, Weiss M, Müller-Höcker M, Lang T, Barros V, Sigmund B, et al. Chronic poisoning by copper in tap water: II. Copper intoxications with predominantly systemic symptoms. Eur J Med Res 1999; 4 (6): 224-8. [ Links ]
96. Sparks DL, Friedland R, Petanceska S, Schreurs BG, Shi J, Perry G, et al. Trace copper levels in the drinking water, but not zinc or aluminum influence CNS Alzheimer-like pathology. J Nutr Health Aging 2006; 10 (4): 247-54. [ Links ]
97. Van Dijk-Looijaard AM, van Genderen J. Levels of exposure from drinking water. Food Chem Toxicol 2000; 38 (1 Suppl): S37-42. [ Links ]
98. Cagnin A, Zambon A, Zarantonello G, Vianello D, Marchiori M, Mercurio D, et al. Serum lipoprotein profile and APOE genotype in Alzheimer’s disease. J Neural Transm Suppl 2007; (72): 175-9.
99. Tabet F, Rye KA. High-density lipoproteins, inflammation and oxidative stress. Clin Sci (Lond). 2009; 116 (2): 87-98. [ Links ]
100. Khera AV, Cuchel M, Llera-Moya M, Amrith Rodrigues MS, Burke MF, Kashif Jafri BA, Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. New Engl J Med 2011; 364: 127-35. [ Links ]
101. Monsuez JJ, Gesquière-Dando A, Rivera S. Cardiovascular prevention of cognitive decline. Cardiol Res Pract 2011; 250970. [ Links ]
102. Nunomura A. Oxidative stress hypothesis for Alzheimer’s disease and its potential therapeutic implications. Rinsho Shinkeigaku 2013; 53 (11): 1043-5.
103. Schrag M, Mueller C, Zabel M, Crofton A, Kirsch WM, Ghribi O, et al. Oxidative stress in blood in Alzheimer’s disease and mild cognitive impairment: a meta-analysis. Neurobiol Dis 2013; 59: 100-10.
104. Lamarche B, Couture P. Dietary fatty acids, dietary patterns, and lipoprotein metabolism. Curr Opin Lipidol 2015; 26 (1): 42-7. [ Links ]
105. Favari E, Chroni A, Tietge UJ, Zanotti I, Escolà-Gil JC, Bernini F. Cholesterol efflux and reverse cholesterol transport. Handb Exp Pharmacol 2015; 224: 181-206. [ Links ]
106. Barone E, Di Domenico F, Butterfield DA. Statins more than cholesterol lowering agents in Alzheimer disease: their pleiotropic functions as potential therapeutic targets. Biochem Pharmacol 2014; 88 (4): 605-16. [ Links ]
107. Levinson SS, Wagner SG. Implications of reverse cholesterol transport: recent studies. Clin Chim Acta 2015; 439: 154-61. [ Links ]
108. Ruiz M, Morosini MLR. Diabetes Mellitus (4a edición). Buenos Aires: Akadia Editorial, 2011. [ Links ]
109. Glorian M, Limon I. Láthérsoclérose, une maladie inflammatoire. Rev Fran Lab 2007; 389: 43-8. [ Links ]
110. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015; 14 (4): 388-405.
111. Herrera MD, Mingorance C, Rodrígue-Rodríguez R, Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Aging Res Rev 2010; 9: 142-52. [ Links ]
112. Joshi YB, Praticó D. The 5-lipoxygenase pathway: oxidative and inflammatory contributions to the Alzheimer’s disease phenotype. Front Cell Neurosci 2015; 8: 436.
113. González-Domínguez R, García-Barrera T, Gómez-Ariza JL. Homeostasis of metals in the progression of Alzheimer’s disease. Biometals 2014; 27 (3): 539-49.
114. Irala J, Martíne-González MA, Guillén-Grima F. ¿Qué es una variable de confusión? Med Clin 2011; 117: 377- 85. [ Links ]
115. Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA 1993; 12-269 (18): 2386-91. [ Links ]
116. Vinyoles Bargalló E, Vila Domènech J, Argimon Pallàs JM, Espinàs Boquet J, Abos Pueyo T, Limón Ramírez E. Concordance among Mini-Examen Cognoscitivo and Mini-Mental State Examination in cognitive impairment screening. Aten Primaria 2002; 15; 30 (1): 5-13. [ Links ]
117. Muñoz-Neira C, Henríquez Ch F, Ihnen JJ, Sánchez CM, Flores MP, Slachevsky Ch A. Psychometric properties and diagnostic usefulness of the Addenbrooke’s Cognitive Examination-Revised in a Chilean elderly sample. Rev Med Chil 2012; 140 (8): 1006-13.
118. Cristalli DO, Arnal N, Marra FA, de Alaniz MJ, Marra CA. Peripheral markers in neurodegenerative patients and their first-degree relatives. J Neurol Sci 2012; 314 (1-2): 48-56. [ Links ]
119. Siritho S, Senanarong V, Nako A, Chotinaiwattarakul W, Jamjumrus P, Udompunthuruk S, et al. Use of Hachinski Ischemic Score in the memory clinic: Thai experience. J Med Assoc Thai 2006; 89 (11): 1822-8. [ Links ]
120. Kim J, Na HK, Byun J, Shin J, Kim S, Lee BH, Na DL.Tracking cognitive decline in amnestic mild cognitive impairment and early-etage Alzheimer dementia: Mini-Mental State Examination versus Neuropsychological Battery. Dement Geriatr Cogn Disord 2017; 44 (1-2): 105-17. [ Links ]
121. Buiza C, Montorio-Cerrato I, Yanguas J. The validity and reliability of the spanish version of the severe cognitive impairment profile (SCIP) for the cognitive assessment of persons with advanced stages of Alzheimer’s disease. Rev Neurol 2015; 16; 61 (8): 349-56.
122. Tamaoka A. Alzheimer’s disease: definition and National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA). Nihon Rinsho 2011; 69 Suppl; 10 Pt 2: 240-5.
123. Beekly DL, Ramos, EM, Van Belle G. The National Alzheimer’s Coordinating Center (NACC) Database: An Alzheimer Disease Database. Alzheimer Dissease Assoc Dis 2004; 18 (4): 270-7.
124. Small GW, Ravins PB, Barry PP, Buckholtz NS, DeKosky ST, et al. Diagnosis and Treatment of Alzheimer Dis Rel Dis, Consensus Statement of the American Association for Geriatric Psychiatry, the Alzheimer’s Association, and the American Geriatrics Society. JAMA 1997; 278 (16): 1363-71.
125. Vargas IT, Fischer DA, Alsina MA, Pavissich JP, Pastén PA, Pizarro GE. Copper corrosion and biocorrosion events in premise plumbing. Materials (Basel) 2017; 5-10 (9). [ Links ]
126. Berkovitch M, Heyman E, Afriat R, Matz-Khromchenko I, Avgil M, Greenberg R, et al. Copper and zinc blood levels among children with nonorganic failure to thrive. Clin Nutr 2003; 22 (2): 183-6. [ Links ]
127. Terrés-Martos C, Navarro-Alarcón M, Martín-Lagos F, López-G de la Serrana H, López-Martínez MC. Determination of copper levels in serum of healthy subjects by atomic absorption spectrometry. Sci Total Environ 1997; 198 (1): 97-103. [ Links ]
128. Bienengräber M, Forderkunz S, Klein D, Summer KH. Determination of Cu-containing metallothionein: comparison of Ag saturation assay, thiomolybdate assay, and enzyme-linked immunosorbent assay. Anal Biochem 1995; 228 (1): 69-73. [ Links ]
129. Ceron JJ, Martinez Subiela S. An automated spectrophotometric method for measuring canine ceruloplasmin in serum. Veterinary Res 2004; 35: 671-9. [ Links ]
130. Prakasam A, Sethupathy S, Lalitha S. Plasma and RBCs antioxidant status in occupational male pesticide sprayers. Clin Chim Acta 2001; 310: 107-12. [ Links ]
131. Twomey PJ, Wierzbicki AS, House IM, Viljoen A, Reynolds TM. Percentage non-caeruloplasmin bound copper. Clin Biochem 2007; 40 (9-10): 749-50. [ Links ]
132. Galland F, Duvillard L, Petit JM, Lagrost L, Vaillant G, Brun JM, Gambert P, Vergès B. Effect of insulin treatment on plasma oxidized LDL/LDL-cholesterol ratio in type 2 diabetic patients. Diabetes Metab 2006; 32 (6): 625-31. [ Links ]
133. Anderson ME, Meister A.Dynamic state of glutathione in blood plasma. J Biol Chem 1980; 255 (20): 9530-3. [ Links ]
134. Benzie IFF, Strain JJ. The ferric reducing ability of plasma as a measure of antioxidant power: the FRAP assay. Anal Biochem 1996; 238: 70-6. [ Links ]
135. Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol 1994; 233: 357-63. [ Links ]
136. Yagi K. A simple fluorometric assay for lipoperoxides in blood plasma. Biochem Med 1976; 15: 121-216. [ Links ]
137. Bradford, MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248-54. [ Links ]
138. Allegri RF, Ollari JA, Mangone CA, Arizaga RL, De Pascale A, Pellegrini M, et al. Grupo de Trabajo de Neuropsicología. Sociedad Neurológica Argentina. El "Mini-Mental State Examination", en la Argentina: Instrucciones para su administración. Rev Neurol Arg 1999; 24 (1): 24-32. [ Links ]
139. J. Butman J, Arizaga RL, Harris P, Drake M, Baumann D, de Pascale A et al. Area de demencia. Test diagnostico. El "Mini - Mental State Examination" en español. Normas para Buenos Aires. Rev Neurol Argent 2001; 26 (1): 11-5. [ Links ]
140. Garau C. "MMSE en la Argentina: instrucciones para su administración"; SNA (1999) en versión anticipada por "Análisis del Mini-Mental State de Folstein en 100 adultos normales" presentada en el XXXIX Congreso Argentino de Neurología (Paraná, Argentina, 1998). [ Links ]
141. Burin DI, Drake MA, Harris P, compiladoras. "Evaluación neuropsicológica en Adultos", Buenos Aires: Editorial Paidós; 2008. [ Links ]
142. El Mini-Mental State Examination: normas para Buenos Aires (editorial). Rev Neurol Argent 2001; 26: 1-15. [ Links ]
143. Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), República Argentina. "Lineamientos para el comportamiento ético del investigador" (2005) y "Lineamientos para el comportamiento en las ciencias biomédicas" (2005). [ Links ]
144. Organización Mundial de la Salud (OMS), "Guías operacionales para Comités de Ética que evalúan Investigación Biomédica", Ginebra, Suiza (2000). [ Links ]
145. Abuja PM, Albertini R. Methods for monitoring oxidative stress, lipid peroxidation and oxidation resistance of lipoproteins. Clin Chim Acta 2001; 306 (1-2): 1-17. [ Links ]
146. Cunqueiro MJ, Comeche MI, Docampo D. Childbirth Self-Efficacy Inventory: psychometric testing of the Spanish version. J Adv Nurs 2009; 65 (12): 2710-8. [ Links ]
147. Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and educational level. J Am Med Assoc 1993; 269(18): 2386-90. [ Links ]
148. Folstein M. Improving dementia assessment by reducing sample heterogeneity. Int Psychogeriatr 2007; 19 (3): 383-9. [ Links ]
149. Batelli S, Albani D, Prato F, Polito L, Franceschi M, Gavazzi A, Forloni G. Early-onset alzheimer disease in an Italian family with presenilin-1 double mutation E318G and G394V. Alzheimer Dis Assoc Disord 2008; 22 (2): 184-7. [ Links ]
150. Urakami K, Wakutani Y, Wada-Isoe K, Yamagata K, Adachi Y, Nakashima K. Causative genes in Alzheimer’s disease. Nihon Ronen Igakkai Zasshi 2001; 38(2): 117-20.
151. Rossi L, Squitti R, Calabrese L, Rotilio G, Rossini PM. Alteration of peripheral markers of copper homeostasis in Alzheimer’s disease patients: implications in aetiology and therapy. J Nutr Health Aging 2007; 11 (5): 408-17.
152. Mangialasche F, Polidori MC, Monastero R, Ercolani S, Camarda C, Cecchetti R, et al. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Res Rev 2009; 8 (4): 285-305.
153. Moreira PI, Sayre LM, Zhu X, Nunomura A, Smith MA, Perry G. Detection and localization of markers of oxidative stress by in situ methods: application in the study of Alzheimer disease. Methods Mol Biol 2010; 610: 419-34. [ Links ]
154. Ventriglia M, Bucossi S, Panetta V, Squitti R. Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimer’s Dis 2012; 30 (4): 981-4.
155. Pearson SD, Ollendorf DA, Colby JA; ICER Alzheimer’s Diagnostics Policy Development Group. Biomarker tests for the diagnosis of Alzheimer’s disease: Generating evidence to inform insurance coverage determinations. Alzheimer’s Dement 2013; 9 (6): 745-52.
156. Puertas MC, Martínez-Martos JM, Cobo MP, Carrera MP, Mayas MD, Ramírez-Expósito MJ. Plasma oxidative stress parameters in men and women with early stage Alzheimer type dementia. Exp Gerontol 2012; 47 (8): 625-30. [ Links ]
157. Chang YT, Chang WN, Tsai NW, Huang CC, Kung CT, Su YJ, et al. The roles of biomarkers of oxidative stress and antioxidant in Alzheimer’s disease: a systematic review. Biomed Res Int 2014: 182303.
158. Bradley-Whitman MA, Lovell MA. Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch Toxicol 2015; 89 (7): 1035-1044. [ Links ]
159. Macreadie IG. Copper transport and Alzheimer’s disease. Eur Biophys J 2008; 37(3): 295-300.
160. Salustri C, Barbati G, Ghidoni R, Quintiliani L, Ciappina S, Binetti G, Squitti RIs cognitive function linked to serum free copper levels? A cohort study in a normal population. Clin Neurophysiol 2010; 121 (4): 502-7. [ Links ]
161. Arnal N, Cristalli DO, de Alaniz MJ, Marra CA. Clinical utility of copper, ceruloplasmin, and metallothionein plasma determinations in human neurodegenerative patients and their first-degree relatives. Brain Res 2010; 1319: 118-30. [ Links ]
162. Inestrosa NC, Cerpa W, Varela-Nallar L. Copper brain homeostasis: role of amyloid precursor protein and prion protein. IUBMB Life 2005; 57 (9): 645-50. [ Links ]
163. Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. Function and regulation of human copper-transporting ATPases. Physiol Rev 2007; 87 (3): 1011-46. [ Links ]
164. Tiffany-Castiglioni E, Hong S, Qian Y. Copper handling by astrocytes: insights into neurodegenerative diseases. Int J Dev Neurosci 2011; 29 (8): 811-8. [ Links ]
165. Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ. Advances in the understanding of mammalian copper transporters. Adv Nutr 2011; 2 (2): 129-37. [ Links ]
166. Hasan NM, Lutsenko S. Regulation of copper transporters in human cells. Curr Top Membr 2012; 69: 137-61. [ Links ]
167. Santos CR, Martinho A, Quintela T, Gonçalves I. Neuroprotective and neuroregenerative properties of metallothioneins. IUBMB Life 2012; 64 (2): 126-35. [ Links ]
168. Dringen R, Scheiber IF, Mercer JF. 2013. Copper metabolism of astrocytes. Front Aging Neurosci 5: 9. [ Links ]
169. Kim H, Wu X, Lee J. 2013. SLC31 (CTR) family of copper transporters in health and disease. Mol Aspects Med 34 (2-3): 561-70. [ Links ]
170. Luo Y, Xu Y, Bao Q, Ding Z, Zhu C, Huang ZX, Tan X. The molecular mechanism for human metallothionein-3 to protect against the neuronal cytotoxicity of Ab(1-42) with Cu ions. J Biol Inorg Chem 2013; 18 (1): 39-47. [ Links ]
171. Inesi G, Pilankatta R, Tadini-Buoninsegni F. Biochemical characterization of P-type copper ATPases. Biochem J 2014; 463 (2): 167-76. [ Links ]
172. Ogra Y. Molecular mechanisms underlying copper homeostasis in Mammalian cells. Nihon Eiseigaku Zasshi 2014; 69 (2): 136-45. [ Links ]
173. Opazo CM, Greenough MA, Bush AI. Copper: from neurotransmission to neuroproteostasis. Front Aging Neurosci 2014; 6: 143. [ Links ]
174. Pal A, Prasad R. Recent discoveries on the functions of astrocytes in the copper homeostasis of the brain: a brief update. Neurotox Rev 2014; 26 (1): 78-84. [ Links ]
175. Scheiber IF, Mercer JF, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol 2014; 116: 33-57. [ Links ]
176. Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, et al. Copper active sites in biology. Chem Rev 2014; 114(7): 3659-853. [ Links ]
177. Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Bio 2012; 4 (5): 1-5.
178. Scheiber I, Dringen R, Mercer JF. Copper: effects of deficiency and overload. Met Ions Life Sci 2013; 13: 359-87. [ Links ]
179. Sidera C, Parsons R, Austen B. Proteolytic cascade in the amyloidogenesis of Alzheimer’s disease. Biochem Soc Trans 2004; 32 (Pt 1): 33-6.
180. Márquez M, Blancas-Mejía LM, Campos A, Rojas L, Castañeda-Hernández G, Quintanar L. 2014. A bifunctional non-natural tetrapeptide modulates amyloid-beta peptide aggregation in the presence of Cu(ii). Metallomics 2014; 6 (12): 2189-92. [ Links ]
181. Migliore L, Uboldi C, Di Bucchianico S, Coppedè F. Nanomaterials and neurodegeneration. Environ Mol Mutagen 2015; 56 (2): 149-170. [ Links ]
182. Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J Mol Neurosci 2001; 17 (2): 137-45.
183. Chopra K, Misra S, Kuhad A. Neurobiological aspects of Alzheimer’s disease. Expert Opin Ther Targets 2011; 15 (5): 535-55.
184. Bieschke J, Zhang Q, Powers ET, Lerner RA, Kelly JW. Oxidative metabolites accelerate Alzheimer’s amyloidogenesis by a two-step mechanism, eliminating the requirement for nucleation. Biochemistry 2005; 44 (13): 4977-83.
185. Lovejoy DB, Guillemin GJ. The potential for transition metal-mediated neurodegeneration in amyotrophic lateral sclerosis. Front Aging Neurosci 2014; 6: 173. [ Links ]
186. Montes S, Rivera-Mancia S, Diaz-Ruiz A, Tristan-Lopez L, Rios C. Copper and copper proteins in Parkinson’s disease. Oxid Med Cell Longev 2014: 147251.
187. De la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem Pharmacol 2014; 88 (4): 548-59.
188. Kalaria RN, Maestre GE, Arizaga R, Friedland RP, Galasko D, Hall K, et al. World Federation of Neurology Dementia Research Group. Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol 2008; 7 (9): 812-26.
189. Hughes TM, Lopez OL, Evans RW, Kamboh MI, Williamson JD, Klunk WE, et al. Markers of cholesterol transport are associated with amyloid deposition in the brain. Neurobiol Ageing 2014; 35 (4): 802-7. [ Links ]
190. Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem 2002; 277(6): 3801-4. [ Links ]
191. Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci 2005; 29 (2): 190-201. [ Links ]
192. Pal A, Prasad R. Recent discoveries on the functions of astrocytes in the copper homeostasis of the brain: a brief update. Neurotox Res 2014; 26 (1): 78-84. [ Links ]
193. Lu J, Zheng YL, Wu DM, Sun DX, Shan Q, Fan SH. Trace amounts of copper induce neurotoxicity in the cholesterol-fed mice through apoptosis. FEBS Lett 200; 580 (28-29): 6730-40. [ Links ]
194. Pocernich CB, Lange ML, Sultana R, Butterfield DA. Nutritional approaches to modulate oxidative stress in Alzheimer’s disease. Curr Alzheimer Res 2011; 8 (5): 452-69.
195. Barnard ND, Bush AI, Ceccarelli A, Cooper J, de Jager CA, Erickson KI, et al. Dietary and lifestyle guidelines for the prevention of Alzheimer’s disease. Neurobiol Aging 2014; 35 Suppl 2: S74-8.
196. Morris MC, Tangney CC. Dietary fat composition and dementia risk. Neurobiol Aging 2014; 35 (Suppl 2): S59-64. [ Links ]
197. Austen BM, Sidera C, Liu C, Frears E. The role of intracellular cholesterol on the processing of the beta-amyloid precursor protein. J Nutr Health Aging 2003; 7 (1): 31-6. [ Links ]
198. Perneczky R, Alexopoulos P, Kurz A. Soluble amyloid precursor proteins and secretases as Alzheimer’s disease biomarkers. Trends Mol Med 2014; 20 (1): 8-15.
199. Arnal N, Astiz M, de Alaniz MJ, Marra CA. Clinical parameters and biomarkers of oxidative stress in agricultural workers who applied copper-based pesticides. Ecotoxicol Environ Saf 2011; 74 (6): 1779-86. [ Links ]
200. De la Cruz D, Cruz A, Arteaga M, Castillo L, Tovalin H. Blood copper levels in Mexican users of the T380A IUD. Contraception 2005; 72 (2): 122-5. [ Links ]
201. Arnal N, de Alaniz MJ, Marra CA. Alterations in copper homeostasis and oxidative stress biomarkers in women using the intrauterine device TCu380A. Toxicol Lett 2010; 192 (3): 373-8. [ Links ]
202. Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys 2004; 423 (1): 37-46. [ Links ]
203. Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 2009; 106 (34): 1467-75.
204. García-Escudero V, Martín-Maestro P, Perry G, Avila J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid Med Cell Longev 2013: 162152. [ Links ]
205. Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 2004; 94 (1): 53-9. [ Links ]
206. Cordy JM, Hooper NM, Turner AJ The involvement of lipid rafts in Alzheimer’s disease. Mol Membr Biol 2006; 23 (1): 111-22.
207. Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis 2008; 29 (3): 422-37.
208. Ramos P, Santos A, Pinto NR, Mendes R, Magalhães T, Almeida A. Anatomical region differences and age-related changes in copper, zinc, and manganese levels in the human brain. Biol. Trace Elem Res 2014; 161 (2): 190-201. [ Links ]
209. Latta CH, Brothers HM, Wilcock DM. Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience 2014; 302: 103-11.
210. Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT. Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 2014; 8: 315. [ Links ]
211. Choo XY, Alukaidey L, White AR, Grubman A. Neuroinflammation and copper in Alzheimer’s disease. Int J Alzheimer’s Dis 2013: 145345.
212. Dusek P, Roos PM, Litwin T, Schneider SA, Flaten TP, Aaseth J. The neurotoxicity of iron, copper and manganese in Parkinson’s and Wilson’s diseases. J Trace Elem Med Biol 2014; 31: 193-203.
213. Dias IH, Polidori MC, Griffiths HR. 2014. Hypercholesterolaemia-induced oxidative stress at the blood-brain barrier. Biochem Soc Trans 42 (4): 1001-5. [ Links ]
214. Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 2010; 345 (1-2): 91-104. [ Links ]
215. Ajith TA, Padmajanair G. Mitochondrial Pharmaceutics: A new therapeutic strategy to ameliorate oxidative stress in Alzheimer’s Disease. Curr Aging Sci 2015; 8 (3): 235-40.
216. Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev 2012; 472932.
217. Ferreira ME, de Vasconcelos AS, da Costa Vilhena T, da Silva TL, da Silva Barbosa A, Gomes AR, et al. Oxidative stress in Alzheimer’s disease: should we keep trying antioxidant therapies?. Cell Mol Neurobiol 2015; 35 (5): 595-614.
218. Rottkamp CA, Nunomura A, Raina AK, Sayre LM, Perry G, Smith MA. Oxidative stress, antioxidants, and Alzheimer disease. Alzheimer Dis Assoc Disord 2000; 14 Suppl 1: S62-6. [ Links ]
219. Huang TC, Lu KT, Wo YY, Wu YJ, Yang YL. Resveratrol protects rats from Ab-induced neurotoxicity by the reduction of iNOS expression and lipid peroxidation. PLoS One 2011; 6 (12): e29102. [ Links ]
220. Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 2003; 189 (1-2): 147-63. [ Links ]
221. Stathopoulou MG, Kanoni S, Papanikolaou G, Antonopoulou S, Nomikos T, Dedoussis G. Mineral intake. Prog Mol Biol Transl Sci 2012; 108: 201-36. [ Links ]
222. Gavrilova SI, Bratsun AL. Epidemiology and risk factors of Alzheimer’s disease. Vestn Ross Akad Med Nauk 1999; (1): 39-46.
223. York SS. IUD litigation: the Planned Parenthood experience. Am J Gynecol Health 1989; 3 (3-S): 35-7. [ Links ]
224. Toyokuni S, Sagripanti JL. Increased 8-hydroxydeoxyguanosine in kidney and liver of rats continuously exposed to copper. Toxicol Appl Pharmacol 1994; 126 (1): 91-7. [ Links ]
225. Babiloni C, Squitti R, Del Percio C, Cassetta E, Ventriglia MC, Ferreri F, et al. Free copper and resting temporal EEG rhythms correlate across healthy, mild cognitive impairment, and Alzheimer’s disease subjects. Clin Neurophysiol 2007; 118 (6): 1244-60.
226. Bhattacharya AK. Global epidemic of stroke--need for urgent intervention. J Indian Med Assoc 2009; 107 (6): 343-4. [ Links ]
227. Roth GA, Fihn SD, Mokdad AH, Aekplakorn W, Hasegawa T, Lim SS. High total serum cholesterol, medication coverage and therapeutic control: an analysis of national health examination survey data from eight countries. Bull World Health Organ 2011; 89 (2): 92-101. [ Links ]
Recibido: 10 de octubre de 2017
Aceptado: 2 de marzo de 2018

