










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkRevista argentina de microbiología
versión impresa ISSN 0325-7541versión On-line ISSN 1851-7617
Rev. argent. microbiol. v.38 n.2 Ciudad Autónoma de Buenos Aires ene./abr. 2006
Mecanismos de acción y de resistencia a rifampicina e isoniacida en Mycobacterium tuberculosis: nueva información sobre viejos conocidos
A. I. De la Iglesia1 Y H. R. Morbidoni*1,2
1Cátedra de Microbiología, Virología y Parasitología, Facultad de Ciencias Médicas, 2Consejo de Investigaciones, Universidad Nacional de Rosario, Santa Fe 3100 (2000) Rosario, Argentina
*Correspondencia. E-mail: hmorbido@unr.edu.ar
RESUMEN
La tuberculosis constituye todavía una de la causas más frecuentes de mortalidad en el mundo. A pesar de la implementación de tratamientos con cuatro drogas antituberculosas, la aparición de cepas resistentes y multirresistentes ha comprometido la eficacia de los mismos. Dos de las drogas en uso, la rifampicina y la isoniacida, recibieron gran atención por su importancia terapéutica, incluso se han identificado los genes involucrados en los mecanismos de resistencia y los que codifican para sus blancos moleculares. La rifampicina es un inhibidor de la subunidad β de la ARN polimerasa de procariotas, incluido Mycobacterium tuberculosis. La resistencia a esta droga está principalmente mediada por mutaciones agrupadas en una región del gen rpoB. Una pequeña fracción de cepas resistentes no mostró mutaciones en rpoB, lo que sugiere la existencia de otros mecanismos de resistencia, posiblemente eflujo de la droga. La isoniacida es una prodroga que se activa por la catalasa-peroxidasa KatG. Mutaciones en katG son las más comúnmente identificadas en cepas clínicas de M. tuberculosis resistentes a isoniacida, confiriendo altos niveles de resistencia. Sin embargo, el blanco molecular de acción para la isoniacida es la InhA, una enoil-ACP reductasa involucrada en la vía de síntesis de los ácidos micólicos. Otras mutaciones involucradas en la resistencia a la isoniacida afectan al gen ndh, que codifica para la NADH deshidrogenasa.
Palabras clave: tuberculosis, resistencia a drogas, mecanismo de acción, isoniacida, rifampicina
ABSTRACT
Mechanisms of action of and resistance to rifampicin and isoniazid in Mycobacterium tuberculosis: new information on old friends. Human tuberculosis is still one of the most frequent causes of death worldwide. Despite the implementation of therapeutic regimes combining four drugs, the rise of resistant and multidrug-resistant Mycobacterium tuberculosis strains has compromised their efficacy. Two of the most effective anti-tubercular drugs in use, rifampicin and isoniazid, have been closely studied due to their therapeutic importance. These studies have led to the identification of the genes involved in resistance mechanisms and of those encoding the molecular targets for these drugs. Rifampicin is an inhibitor of the β-subunit of the RNA polymerase of prokaryotes, including M. tuberculosis. Resistance to rifampicin is mediated by mutations clustered in a small region of the rpoB gene. A fraction of resistant strains showed no mutations in rpoB, suggesting that other mechanisms of resistance, possibly efflux pumps, may exist. Isoniazid is a pro-drug activated by KatG, a catalase-peroxidase. Mutations in katG, the most commonly found in M. tuberculosis clinical isolates, give high levels of resistance. In spite of this, the molecular target for isoniazid is InhA, an enoyl-ACP-reductase involved in the biosynthesis of mycolic acids. Other mutations causing resistance to isoniazid have been mapped to ndh, a gene encoding the NADH dehydrogenase.
Key words: tuberculosis, drug resistance, mechanisms of action, isoniazid, rifampicin
INTRODUCCIÓN
La tuberculosis humana sigue siendo una de las causas principales de muerte en el mundo y su tratamiento es todavía una verdadera batalla de pronóstico incierto (26). El escaso arsenal de drogas antituberculosas disponibles no ha tenido adiciones de nuevos fármacos antituberculares específicos en los últimos 30 años (47), por lo que el éxito del tratamiento depende en gran medida de su estricto cumplimiento (93, 101). La mejora en los resultados de la terapia antituberculosa se ha debido, fundamentalmente, a la implementación del sistema DOTS (Directly Observed Treatment, Short Course, o tratamiento breve directamente observado), basado en la toma de las drogas antituberculosas por parte del paciente, supervisado por personal de salud para asegurar el cumplimiento del tratamiento (36, 74). La quimioterapia contra M. tuberculosis es diferente a la utilizada contra otros patógenos bacterianos debido al largo tiempo de duplicación de esta micobacteria (18-24 horas, comparado con los 30-40 minutos requeridos por otras bacterias), a su constitución estructural, que le confiere una baja permeabilidad celular (56, 73, 90), y a su capacidad de permanecer en el huésped en estado de latencia (140). También es importante destacar que este patógeno puede ser hallado en distintas ubicaciones dentro del huésped, donde enfrenta distintas condiciones ambientales (82, 83). De esta manera, podemos pensar en la enfermedad activa como la suma de distintas poblaciones dinámicas en el organismo. Así, dado que los pulmones son los órganos más frecuentemente afectados, en ellos pueden encontrarse micobacterias dividiéndose activamente o manifestando un metabolismo reducido en las lesiones caseosas, en condiciones de bajo pH y bajas tensiones de oxígeno. Estas poblaciones son metabólicamente distintas y es razonable esperar diferente susceptibilidad a drogas antituberculosas, como fue postulado por D. A. Mitchison (82, 83). Un modelo matemático desarrollado por Lipsitch y Levin tomó en cuenta esta hipótesis y llevó a la conclusión de que el abandono o la irregularidad del tratamiento eran factores más importantes que la heterogeneidad poblacional micobacteriana (72).
Las razones mencionadas explican la necesidad de instaurar una terapia basada en la administración de cuatro fármacos bajo el sistema DOTS, a fin de llegar a las distintas poblaciones de micobacterias para reducir drásticamente su número y minimizar la aparición de mutantes resistentes a las drogas.
En el tratamiento antituberculoso estandarizado se utilizan cuatro drogas, rifampicina (RIF), isoniacida (INH), etambutol (EMB) y pirazinamida (PZA), durante la primera fase de dos meses (llamada intensiva), la cual se continúa con una segunda fase de cuatro meses con dos drogas (RIF e INH) (83, 84). Como se ve, RIF e INH forman parte de ambas fases y constituyen las drogas más importantes del tratamiento. La resistencia a ambos agentes antituberculosos obliga a elegir drogas de menor efectividad, mayor toxicidad y mayor costo en su reemplazo, denominadas drogas de segunda línea. De lo descrito se desprende lo primordial de determinar si una cepa en estudio es resistente a RIF, a INH o a ambos antibióticos, ya que esta situación determinará en gran medida el éxito del tratamiento y la complejidad y costo del manejo terapéutico (34, 36, 37).
Las herramientas genéticas desarrolladas a partir de 1990 (16, 38, 55, 105) han permitido el análisis de los mecanismos de resistencia a algunas de las drogas antituberculosas. Dado el valor terapéutico de ambas, la rifampicina y la isoniacida fueron las primeras drogas cuyos mecanismos de acción y de resistencia fueron estudiados a nivel molecular. El aislamiento de mutantes resistentes a RIF o INH en el laboratorio ha permitido el estudio de los genes más frecuentemente afectados por mutaciones, las cuales han sido validadas al ser detectadas en cepas clínicas resistentes a estas drogas. En las secciones siguientes se describe el conocimiento actual de los mecanismos de acción de la RIF y la INH, así como los mecanismos involucrados en la resistencia a estos antibióticos.
Bases genéticas de la resistencia a rifampicina
Ha sido claramente demostrado en Escherichia coli que la rifampicina actúa inhibiendo la síntesis de ARN mensajero, a cargo de la ARN polimerasa (38). Asimismo, mutantes de esta enterobacteria adquirían resistencia a RIF mediante mutaciones en rpoB, el gen codificante para la subunidad β de la ARN polimerasa. Basados en estos indicios, Telenti et al. clonaron el gen rpoB de M. tuberculosis, determinaron su secuencia y usaron esta información para amplificar dicho gen en 54 aislamientos clínicos RIFR y en 66 RIFS (128). Estos investigadores hallaron que todas las cepas resistentes tenían mutaciones en rpoB, mientras que las cepas sensibles no presentaban mutaciones en este gen. Las mutaciones causantes de resistencia a RIF en M. tuberculosis estaban agrupadas en una pequeña zona de rpoB e involucraban sólo 8 de los 23 aminoácidos presentes en esa región, especialmente en los residuos 526 y 531 (127, 128). Estos datos fueron corroborados y extendidos mediante el análisis de numerosos aislamientos clínicos en distintos países. Se describieron mutaciones puntuales, inserciones y deleciones que afectan una región de 100 pb en casi la totalidad (95%) de los casos estudiados (88, 89, 100). Aprovechando las técnicas de manipulación de ADN existentes y sobre la base del conocimiento reunido acerca de las mutaciones en rpoB, Kapur et al. secuenciaron 128 cepas clínicas de EE.UU., entre las que se incluyen 121 cepas RIFR (59). Estos investigadores detectaron polimorfismos en la zona correspondiente a los 23 residuos de aminoácidos mencionados; se cuentan entre ellos dos inserciones de tres o seis bases, tres deleciones de seis y nueve bases y seis mutantes con dos mutaciones puntuales en esta zona del gen rpoB. Nuevos estudios realizados por una gran cantidad de laboratorios reportan las mismas mutaciones, hecho que valida estos hallazgos, (1, 2, 4, 5, 19, 23, 42, 44, 52, 53, 67, 112, 119).
En un reporte reciente, Cummings y Segal analizaron la frecuencia de cambios de aminoácidos que tenían una incidencia marcada sobre los valores de concentración inhibitoria mínima (CIM) para RIF (27). Para ello elaboraron un modelo de análisis de los cambios en la secuencia de la zona 511-533 de rpoB de diversos aislamientos clínicos, y los correlacionaron con los valores de CIM. Las mutaciones implicadas en el cambio de aminoácidos fueron halladas en las posiciones 511, 513, 515, 516, 521, 526, 531 y 533; sin embargo, las mutaciones relevantes que causaban aumento de la CIM para RIF correspondían solamente a los residuos 526 y 531, con incrementos de entre 100 y 400 veces (Tabla 1). Por el contrario, cambios polimórficos en los residuos 511, 512, 515, 521 y 529 apenas tenían incidencia en los niveles de CIM (27). Este análisis indica que la secuenciación de un fragmento de rpoB puede ser una manera rápida de determinar el nivel de resistencia a RIF de un aislamiento, en reemplazo de la más lenta técnica clásica de cultivo (18, 79, 80, 117, 139).
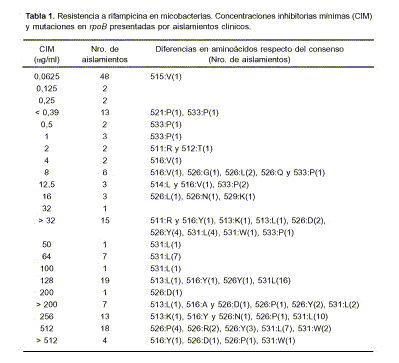
Si bien no hay duda de que mutaciones en una región pequeña de rpoB son responsables de la resistencia a rifampicina, no todas ellas confieren resistencia a las demás rifamicinas de uso clínico. La rifabutina es una rifamicina utilizada como droga de segunda línea en el tratamiento de infecciones por M. tuberculosis y M. avium. Mutaciones en los codones 526 y 531 de rpoB causan resistencia a la rifampicina y a la rifabutina (94, 116); sin embargo, las restantes mutaciones descritas en esta sección determinan resistencia a la rifampicina pero no a otras rifamicinas, como lo demostraron Anthony et al. (7). Estos autores también demostraron que la frecuencia de aparición de mutantes resistentes a la rifabutina es 10 veces mayor cuando se obtienen a partir de mutantes resistentes a la rifampicina, comparada con la frecuencia de aparición de mutantes cuando se selecciona directamente por resistencia a la rifabutina, sin selección previa para rifampicina. (34, 54, 59, 114).
A pesar de haberse identificado claramente a rpoB como el gen más frecuentemente asociado a la resistencia a RIF en micobacterias, se ha reportado un pequeño número de cepas resistentes sin mutaciones en este gen. Es importante considerar la posibilidad de mecanismos de resistencia alternativos, como son las bombas de eflujo, encargadas de eliminar una gran variedad de compuestos del citoplasma de las bacterias (71, 114).
Resistencia a rifampicina y concepto de fitness bacteriano
RIF e INH son drogas esenciales en el tratamiento de la tuberculosis; el aislamiento de cepas clínicas resistentes a estas drogas ha creado un gran interés en la frecuencia de aparición de dichos mutantes y en la identificación y el análisis de los genes involucrados en los mecanismos de resistencia. También resultó de interés estudiar el efecto que estas mutaciones ocasionan en la fisiología celular. Este concepto, desarrollado para otros géneros bacterianos, es conocido como bacterial fitness (28, 46, 62, 77, 118). El mismo postula que la reducción en el uso de drogas antimicobacterianas causaría la desaparición de las cepas resistentes debido a que toda mutación tendría un costo adverso en la fisiología de la célula mutante, traducido en una velocidad de crecimiento menor comparada con las células salvajes. Esto llevaría al desplazamiento de las células mutantes en la población total de micobacterias. M. tuberculosis muestra una amplia variedad de fenotipos medibles de fitness, incluidos tiempo de duplicación, transmisibilidad, infectividad y patogenicidad, que podrían significar diferencias in vivo (131). Estudios recientes han añadido más polémica al tema, al demostrar que la medición del fitness puede variar según el ensayo utilizado y, más aún, que pueden obtenerse distintos resultados si se comparan valores obtenidos in vitro con los obtenidos in vivo. Al mismo tiempo, los costos ocasionados por las mutaciones que confieren resistencia a drogas pueden ser minimizados por mutaciones compensatorias, sin pérdida de la resistencia y sin afectar mayormente la capacidad de crecimiento (15, 28, 46, 62, 118). Estas mutaciones pueden ser de costo mínimo para las células, como demostraron Sander et al. mediante un modelo en el que utilizan la micobac-teria no patógena M. smegmatis (110). Estos hechos sugieren que, lejos de desaparecer, las mutantes resistentes pueden persistir en la población. Es por ello que es necesario estudiar la velocidad de aparición de mutantes, así como también los efectos de las mutaciones sobre el ciclo celular. Un ejemplo de esto es la reciente publicación de Mariam et al. (77), donde se reporta el aislamiento in vitro de mutantes RIFR. La secuenciación de rpoB en estos mutantes mostró mayoritariamente la presencia de las mutaciones Ser531Trp, His526Tyr y Ser522Leu, con valores de CIM superiores a 32 µg/ml. Estos autores determinaron los tiempos de duplicación para la cepa salvaje y las cepas mutantes, la capacidad de crecimiento en macrófagos y el fitness relativo (tiempo de duplicación de la cepa salvaje/ tiempo de duplicación de la cepa mutante) en ensayos de cultivo puro y de competencia con la cepa salvaje (77). Los valores hallados, mostrados en la Tabla 2, ejemplifican el efecto que las mutaciones ejercen sobre la capacidad metabólica celular. Por ejemplo, la mutante Harlingen rpoB Ser531Trp ha sido aislada de pacientes con baja frecuencia (14% del total de aislamientos), lo que podría atribuirse al bajo fitness de esta mutante in vivo. En cambio, los reemplazos Ser531Leu e His526Tyr en el gen rpoB de la misma cepa presentan una mayor capacidad de crecimiento in vivo (0,84), lo que explica su mayor prevalencia de aislamiento (12).
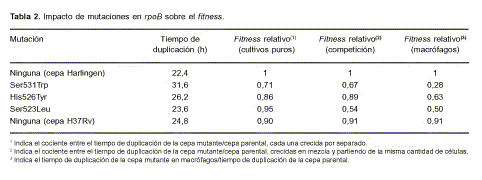
Si bien el análisis de impacto de la resistencia a INH sobre el fitness de M. tuberculosis es un tópico de evidente importancia, el mismo deberá aguardar hasta la identificación de todos los genes involucrados en la resistencia y la comprensión del rol celular de los mismos.
Resistencia a isoniacida en micobacterias: mutantes deficientes en catalasa-peroxidasa (KatG) son resistentes a la isoniacida
La isoniacida (forma hidracida del ácido isonicotínico) es una droga de gran actividad sobre M. tuberculosis, con una CIM de 0,05 µg/ml. Trabajos pioneros de F. Winder (139-149) y su posterior expansión por K. Takayama (120-124), demostraron que INH inhibía específicamente la síntesis de los ácidos micólicos, sin afectar la síntesis de otros ácidos grasos. Los ácidos micólicos son ácidos grasos ramificados de cadena muy larga (hasta 90 átomos de carbono), constituyen uno de los componentes principales de la envoltura de la célula micobacteriana y contribuyen, en gran medida, a su impermeabilidad (73). Sin embargo, se debió esperar hasta los años noventa para que los avances en la manipulación genética de micobacterias permitieran el clonado y la expresión de los genes responsables de la resistencia a INH. Uno de los primeros avances en este sentido lo constituyó la dilucidación del rol en la actividad an-tituberculosa de INH de la enzima KatG, una catalasa-peroxidasa. Así, Zhang et al. demostraron que INH era, en realidad, una prodroga que se activaba por KatG, lo que da origen a una variedad de especies reactivas (151, 152). Posteriormente, trabajos de Heym et al. identificaron katG en el cromosoma de varias micobacterias, entre ellas M. tuberculosis, y estudiaron además las mutaciones que afectaban a este gen en los aislamientos clínicos INHR (49, 51). Hasta el presente, una larga serie de reportes indican que katG es responsable de casi el 60% de los casos de INHR en cepas aisladas de pacientes (3, 34, 39, 45, 49, 53, 54, 78, 109, 110, 134, 152). La mutación más comúnmente observada en katG es en el codón 315, que causa el cambio del aminoácido Serina 315 por Treonina (Ser315Thr), y está asociada con altos niveles (5-10 µg/ml) de resistencia a INH (53, 78). La información generada ha servido, como en el caso de la resistencia a RIF, para identificar las mutaciones en katG responsables de la resistencia, lo que ha permitido diseñar sistemas de detección basados en la secuenciación de ADN, en la reacción en cadena de la polimerasa (PCR) o en polimorfismos de cadena simple de ADN (SSCP) (33, 34, 54, 59, 61, 76). Estos estudios son de gran valor para determinar rápidamente si una cepa clínica puede ser INHR debido a la pérdida de la activación de INH ya que, como se ha señalado, las mutaciones en katG son las más comunes. Sin embargo, la existencia de otros genes implicados en la resistencia (como se describe en las próximas secciones) deja una fracción importante de aislamientos clínicos en los que las técnicas de PCR no son aplicables para determinar la resistencia o sensibilidad a INH, al no conocerse la identidad de los genes mencionados.
Otro interesante punto para remarcar es que katG es un gen no esencial y, por lo tanto, no se ajusta a la definición de blanco de acción de una droga. Entonces, la pregunta es: ¿cuál(es) es(son) la(s) enzima(s) inhibida(s) por la forma activada de INH en micobacterias y cuántos genes están involucrados en la resistencia a esta droga?
La isoniacida inhibe a la enoil-ACP reductasa, enzima requerida para la síntesis de ácidos micólicos
Una de las mayores sorpresas deparadas por la secuenciación del genoma de M. tuberculosis fue la observación de la presencia de gran cantidad de genes dedicados a la degradación, síntesis y modificación de ácidos grasos (24). Uno de los genes presentes en el cromosoma de este patógeno (así como también en el de otras especies de micobacterias) codifica para una enzima que contiene todos los dominios funcionales requeridos para la síntesis de ácidos grasos, denominada sintetasa de ácidos grasos de tipo I o FASI (Fatty Acid Synthase I). Lo llamativo de la presencia de esta enzima en micobacterias es que es la misma que sintetiza ácidos grasos en eucariotas, desde la levadura Saccharomyces cereviseae hasta el ser humano (20, 57, 63, 115, 137). Estudios realizados por K. Bloch en la década del setenta mostraron una actividad enzimática de alto peso molecular presente en M. smegmatis, capaz de sintetizar ácido palmítico in vitro (13, 14, 92, 96). Esto es sorprendente si se considera que en la gran mayoría de los géneros bacterianos, la síntesis de ácidos grasos procede a través de las mismas reacciones químicas pero catalizadas por varias enzimas (o sea, por distintas proteínas), cada una de las cuales lleva a cabo una etapa de la reacción de síntesis (75, 107). Este conjunto de proteínas, físicamente independientes, se asocian para llevar a cabo las reacciones de síntesis y recibe la denominación de sintetasa de ácidos grasos de tipo II o FASII (Fatty Acid Synthase II). Como se ha mencionado en la sección precedente, trabajos publicados por Winder (139-149) y por Takayama (122-124) demostraron que INH inhibía la síntesis de ácidos micólicos. Sin embargo, no se conocían cuáles eran las enzimas implicadas en dicha síntesis. La identificación del blanco de acción de INH en micobacterias fue realizada, en su mayor parte, por el grupo de W.R. Jacobs, y contribuyó no solamente a aclarar el mecanismo de acción de la isoniacida sino también a comprender la ruta biosintética de los ácidos micólicos, como se describe a continuación.
El grupo de Jacobs aisló cepas mutantes de la mico-bacteria no patógena de crecimiento rápido M. smegmatis resistentes a INH, y clonó e identificó el gen responsable (8). Este gen, también presente con alto grado de homología en M. tuberculosis y M. bovis BCG, fue denominado inhA y se vio que mostraba gran similitud con el gen fabI de E. coli, involucrado en la síntesis de ácidos grasos de bacterias mediada por FASII (Figura 1). Esta información adquiere relevancia si se recuerda que INH causa inhibición de la síntesis de los ácidos micólicos en micobacterias, pero no afecta la síntesis de los ácidos grasos de cadena más corta producidos por FASI. Al mismo tiempo podría estar señalando la existencia de dicho sistema en micobacterias, dado que fabI es parte de FASII. Esta posibilidad estaría sustentada por la necesidad de contar con un sistema de síntesis para los ácidos micólicos, con lo cual las micobacterias tendrían sistemas específicos para la síntesis de ácidos grasos “normales” (necesarios para la síntesis de membrana citoplasmática) y “extra-largos”.
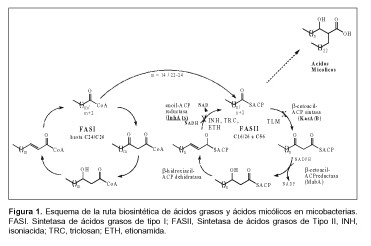
La secuenciación del gen mutado en cepas de M. smegmatis INHR mostró que el cambio del aminoácido Serina 94 por Alanina (Ser94Ala) era el responsable de la resistencia a INH. Asimismo se demostró que esta cepa mutante presentaba resistencia cruzada a la droga antuberculosa de segunda línea etionamida (8), la que también actúa inhibiendo la síntesis de ácidos micólicos (97). Sin embargo, numerosos aislamientos clínicos INHR eran ETHS, lo que sugiere blancos distintos. Estudios llevados a cabo por Ortiz de Montellano y otros investigadores mostraron recientemente que la etionamida es también una prodroga activada por una monooxigenasa denominada EthA (10, 31, 131), y esto justificaría la proporción de aislamientos clínicos INHR-ETHS. El hecho de que cepas clínicas mostraran resistencia tanto a INH como a ETH indicaba un mecanismo de resistencia compartido, lo que fue confirmado al clonar y secuenciar el gen inhA en cepas INHR katG (+) y ETHR ethA (+). En todas las cepas analizadas se encontraron mutaciones en inhA que eran suficientes para impedir la unión de INH* (INH activado) y ETH* (ETH activado) (48, 65, 69, 86, 106).
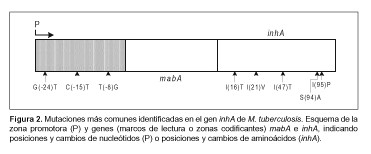
La identificación de InhA como blanco de acción de INH en micobacterias llevó a la secuenciación del gen inhA en numerosos aislamientos clínicos, y se encontró que las mutaciones más frecuentes eran de dos tipos: 1) en la región promotora de inhA (este gen está ubicado corriente abajo del gen mabA, con el cual forma un operón) y 2) en la región codificante de inhA. Dos trabajos muy completos en cuanto a la naturaleza de las mutaciones detectadas en katG e inhA son los de Morlock et al. (86) y Cardoso (17), donde se analizan cepas clínicas de EE.UU., Rusia y Brasil. Las mutaciones más frecuentemente halladas en las zonas promotora y codificante de mabA-inhA se muestran en la Figura 2. En todos los casos estudiados, las mutaciones que afectaban inhA otorgaban bajos niveles de resistencia a INH, en contraposición a la alta resistencia mediada por mutaciones en katG. En cierta manera es lo esperable, dado que InhA es una enzima esencial y, por lo tanto, sólo puede soportar una cierta cantidad de reemplazos de aminoácidos sin alterar su función enzimática. En cambio KatG es una enzima no esencial y su pérdida impide la activación de INH. En este contexto es interesante comentar que mientras M. tuberculosis INHR acepta relativamente pocas mutaciones en el marco de lectura y zona del promotor del gen inhA (Figura 2), M. smegmatis INHR, que ha sido usado como un microorganismo modelo, tolera muchas más mutaciones en dicho gen (89). Dado que ambos genes son conservados, estos resultados sugieren que la expresión y actividad de la enoil-ACP reductasa está más estrictamente controlada en M. tuberculosis (H.R. Morbidoni et al., manuscrito en preparación).
Trabajos posteriores llevados a cabo por Jacobs y sus asociados incluyeron la caracterización bioquímica y biofísica de la enzima InhA, y comprobaron que la forma activada de INH (INH*) se unía a NADH (cofactor de la reacción catalizada por InhA) y luego el complejo reaccionaba con InhA causando su inactivación (9, 32, 98).
Para validar el rol de InhA como enzima esencial y blanco de acción de INH, Vilcheze et al. aislaron mutantes termosensibles (TS) de M. smegmatis resistentes a INH (133, 134). Una de estas mutantes fue caracterizada y se encontró que la mutación que causaba la resistencia a INH y la termosensibilidad afectaba a InhA, por lo tanto quedaba inequívocamente demostrado que inhA es un gen esencial y que es el blanco de acción de la forma activada de INH. Cultivos en fase de crecimiento exponencial de esta mutante cambiados a temperaturas no permisivas, mostraban inhibición de la síntesis de ácidos micólicos y cinética de lisis indistinguible de cultivos salvajes de M. smegmatis o M. bovis var. BCG tratados con INH (135). Conjuntamente con esos resultados, Larsen et al. demostraron que la sobrexpresión de inhA era suficiente para incrementar la CIM para INH y ETH en M. tuberculosis, M. bovis var BCG y M. smegmatis, lo que prueba que InhA es el blanco de acción de ambas drogas antituberculosas (64). Estos resultados son de importancia ya que ETH es una droga de segunda línea que se usa en reemplazo de INH cuando la cepa es INHR; como se ve, ETH puede ser utilizada si se comprueba que la resistencia a INH es causada por mutaciones en katG.
El mecanismo de acción de INH involucra otros genes desconocidos, ya que se han aislado cepas clínicas resistentes a INH que no presentaban mutaciones en katG ni en inhA (66, 68). Estudios llevados a cabo en el laboratorio del Dr. W. R. Jacobs identificaron un nuevo gen involucrado en la resistencia a INH, ndh, que codifica para la enzima NADH deshidrogenasa (81). Las mutantes aisladas mostraban corresistencia a INH y ETH, con valores de CIM de 2 a 6 veces más altos que los de la cepa salvaje (M. bovis BCG var. Pasteur) (136). Este resultado es esperable dado que, como se mencionó anteriormente, la forma activada de INH se une a una molécula de NADH (cofactor de la reacción) para inhibir a InhA (32, 99). Mutaciones en el gen ndh fueron descritas en cepas clínicas, hecho que valida los estudios basados en mutantes espontáneas de M. bovis BCG resistentes a INH con mutaciones en dicho gen, aisladas en el laboratorio (68).
La Tabla 3 resume el conocimiento actual de los genes involucrados en la resistencia a la isoniacida, así como también los valores de CIM observados en cada caso. El rol de otros genes (como los que participan en la regulación de la expresión de katG y que, por lo tanto, influyen en el nivel de actividad de KatG en la célula) queda fuera de los alcances de esta revisión y será considerado en futuras publicaciones. Los mecanismos de acción identificados o propuestos para INH se muestran esquematizados en la Figura 3.

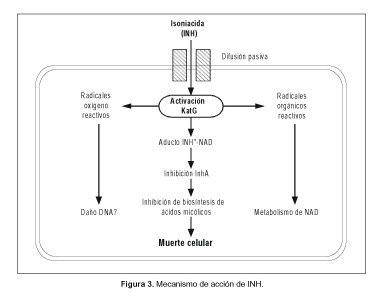
En resumen, tanto INH como ETH son prodrogas activadas por la catalasa-peroxidasa KatG y la monooxi-genasa EthA, respectivamente. Ambas prodrogas, luego de su activación, inhiben a InhA, la enoil-ACP reductasa involucrada en la síntesis de ácidos micólicos, lo que explica la aparición de cepas clínicas resistentes a ambas drogas antituberculosas. La información generada sobre el mecanismo de acción y de resistencia a INH y ETH debe ser tenida en cuenta al identificarse cepas clínicas de M. tuberculosis resistentes a INH, por la probabilidad de que también presenten resistencia cruzada a ETH.
Diagnóstico de resistencia a isoniacida: ¿termociclar o no termociclar?
Si se considera el tiempo de duplicación de M. tuberculosis (18-24 h) y el tiempo de aparición de colonias en medios de cultivo sólidos (de uno a dos meses), la determinación de la sensibilidad o resistencia a las drogas antituberculosas difícilmente se pueda informar al infectólogo tratante en menos de dos meses. Es clara la necesidad de contar con metodologías que permitan realizar estos estudios en un lapso sensiblemente menor, para evitar la instauración de tratamientos empíricos que quizás no sólo resulten inefectivos sino que, tal vez, favorezcan la propagación de cepas resistentes a drogas en la comunidad.
La utilización de la reacción en cadena de la polimerasa (PCR) constituyó uno de los mayores avances en las ciencias biológicas en los últimos veinte años. Una de sus consecuencias más visibles ha sido la aplicación de técnicas de amplificación de material genético para revelar la presencia de diversos microorganismos (virus, bacterias, hongos) en muestras biológicas, aun presentes en muy baja cantidad, lo que permite un diagnóstico rápido y sensible (6, 40, 70, 120). De la misma manera, la amplificación de secuencias específicas (junto con otras técnicas fenotípicas de detección), ha permitido evidenciar la presencia de genes que codifican enzimas involucradas en mecanismos de patogénesis, tales como la producción de toxinas (11, 60, 87) o la resistencia a antibióticos (25, 29, 35, 43, 91, 102, 113). Los mecanismos de resistencia a drogas en micobacterias involucran genes cromosomales (50), a diferencia de otras especies donde varios de los mecanismos de resistencia están codificados en plásmidos, como ocurre con las β-lactamasas (58, 95, 103, 108, 121). En estos casos, los genes que causan la resistencia a antibióticos son mucho más fácilmente evidenciables, ya que no tienen un correlato cromosomal y su detección implica el carácter de resistencia (104, 112). En comparación con otros grupos, la determinación de la resistencia a drogas por métodos moleculares en micobacterias no es tan fácil y depende de la cantidad de genes cromosomales implicados. Un ejemplo, basado en lo comentado en las secciones precedentes, es la resistencia a RIF, donde la mayoría de las mutaciones se concentran en una pequeña zona del gen, de relativamente fácil amplificación y análisis (21, 30, 85). Sin embargo, si bien las técnicas de amplificación de ADN se utilizan para demostrar la resistencia a RIF, no son sencillas de aplicar en la determinación de resistencia a INH. Esto es debido a que la resistencia a INH requeriría el análisis de varios genes, aun teniendo en cuenta que la mayoría de las mutaciones deberían afectar a katG, que contienen un número variable de mutaciones, no siempre agrupadas dentro del gen. Sin embargo, se han publicado trabajos donde se reporta la utilización de métodos moleculares para la determinación rápida de resistencia a INH, la mayoría de los cuales son en general costosos y requieren personal entrenado y equipamiento de complejidad media o alta (22, 33, 41, 104, 129, 130, 132).
Resistencia a RIF: ¿marcador presuntivo de resistencia a INH?
Uno de los misterios pendientes de resolución en esta historia es la frecuente aparición simultánea de resistencia a RIF y a INH. Dado que ambas drogas son la columna vertebral del tratamiento antituberculoso, es de vital importancia conocer a la brevedad si la cepa en estudio es resistente o sensible a estos antibióticos. Desafortunadamente para el éxito del tratamiento terapéutico, la literatura basada en los casos clínicos analizados reporta la resistencia a RIF como un marcador de resistencia a INH. Si bien los datos experimentales avalan la teoría según la cual las mutaciones causales de resistencia a RIF pueden disminuir la capacidad transcripcional de las mutantes (15, 28), hasta el momento se desconoce la razón capaz de explicar por qué cepas RIFR son muy frecuentemente INHR . Es de tener en cuenta el modelo matemático sugerido por Lipsitch y Levin (72), en el cual la existencia de distintos compartimentos con micobac-terias en distinto estado metabólico (y por consiguiente, con distinta sensibilidad a drogas) explicaría la posibilidad de aparición de mutantes resistentes a una droga, y en estadios posteriores, de resistencia secundaria. En este análisis, la resistencia a INH, la droga más potente, aparece y se convierte en predominante en la población cuando no hay adherencia al tratamiento.
Agradecimientos: los autores agradecen la excelente labor secretarial del Sr. Adrián Fornasari. HRM es Investigador Independiente del CIUNR (Consejo de Investigaciones de la Universidad Nacional de Rosario).
REFERENCIAS
1. Agdamag DM, Kageyama S, Solante R, Espantaleon AS, Sangco JC, Suzuki Y. Characterization of clinical isolates of Mycobacterium tuberculosis resistant to drugs and detection of RpoB mutation in multidrug-resistant tuberculosis in the Philippines. Int J Tuberc Lung Dis 2003; 7: 1104-8. [ Links ]
2. Ahmad S, Araj GF, Akbar PK, Fares ET, Chugh D, Mustafa AS. Characterization of rpoB mutations in rifampin-resistant Mycobacterium tuberculosis isolates from the Middle East. Diagn Microbiol Infect Dis 2000; 38: 227-32. [ Links ]
3. Ahmad S , Mokaddas E. Contribution of AGC to ACC and other mutations at codon 315 of the katG gene in isoniazid-resistant Mycobacterium tuberculosis isolates from the Middle East. Int J Antimicrob Agents 2004; 23: 473-9. [ Links ]
4. Ahmad S, Mokaddas E, Fares E. Characterization of rpoB mutations in rifampin-resistant clinical Mycobacterium tuberculosis isolates from Kuwait and Dubai. Diagn Microbiol Infect Dis 2002; 44: 245-52. [ Links ]
5. Aktas E, Durmaz R, Yang D, Yang Z. Molecular characterization of isoniazid and rifampin resistance of Mycobacterium tuberculosis clinical isolates from Malatya, Turkey. Microb Drug Resist 2005; 11: 94-9. [ Links ]
6. Al-Bakkal G, Ficarra G, McNeill K, Eversole LR, Sterrantino G, Birek C. Human papilloma virus type 16 E6 gene expression in oral exophytic epithelial lesions as detected by in situ rtPCR. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1999; 87: 197-208. [ Links ]
7. Anthony RM, Schuitema AR, Bergval IL, Brown TJ, Oskam L, Klatser PR. Acquisition of rifabutin resistance by a rifampicin resistant mutant of Mycobacterium tuberculosis involves an unusual spectrum of mutations and elevated frequency. Ann Clin Microbiol Antimicrob 2005; 4: 9-15. [ Links ]
8. Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, et al. InhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994; 263: 227-30. [ Links ]
9. Basso LA, Zheng R, Musser JM, Jacobs Jr WR, Blanchard JS. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis: enzymatic characterization of enoyl reductase mutants identified in isoniazid-resistant clinical isolates. J Infect Dis 1998; 178: 769-75. [ Links ]
10. Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, et al. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem 2000; 275: 28326-31. [ Links ]
11. Berry C, O’Neil S, Ben-Dov E, Jones AF, Murphy L, Quail MA, et al. Complete sequence and organization of pBtoxis, the toxin-coding plasmid of Bacillus thuringiensis subsp. israelensis. Appl Environ Microbiol 2002; 68: 5082-95. [ Links ]
12. Billington OJ, McHugh TD, Gillespie SH. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 1999; 43: 1866-9. [ Links ]
13. Bloch K. Fatty acid synthases from Mycobacterium phlei. Methods Enzymol. 1975; 35: 84-90. [ Links ]
14. Bloch K. Control mechanisms for fatty acid synthesis in Mycobacterium smegmatis. Adv Enzymol Relat Areas Mol Biol 1977; 45: 1-84. [ Links ]
15. Bottger EC, Springer B, Pletschette M, Sander P. Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nat Med 1998; 4: 1343-4. [ Links ]
16. Braunstein M, Bardarov SS, Jacobs Jr WR. Genetic methods for deciphering virulence determinants of Mycobacterium tuberculosis. Methods Enzymol 2002; 358: 67-99. [ Links ]
17. Cardoso RF, Cooksey RC, Morlock GP, Barco PL, Cecon F, Forestiero C, et al. Screening and characterization of mutations in isoniazid-resistant Mycobacterium tuberculosis isolates obtained in Brazil. Antimicrob Agents Chemother 2004; 48: 3373-81. [ Links ]
18. Carvalho WS, Spindola MS, de Costa KM, Araujo JG, Augusto CJ, Pesquero JB, et al. Low-stringency single-specific-primer PCR as a tool for detection of mutations in the rpoB gene of rifampin-resistant Mycobacterium tuberculosis. J Clin Microbiol 2003; 41: 3384-6. [ Links ]
19. Cavusoglu C, Hilmioglu S, Guneri S, Bilgic A. Characterization of rpoB mutations in rifampin-resistant clinical isolates of Mycobacterium tuberculosis from Turkey by DNA sequencing and line probe assay. J Clin Microbiol 2002; 40: 4435-8. [ Links ]
20. Chirala SS, Wakil SJ. Structure and function of animal fatty acid synthase. Lipids 2004; 39: 1045-53. [ Links ]
21. Cirillo DM, Piana F, Frisicale L, Quaranta M, Riccabone A, Penati VV, et al. Direct rapid diagnosis of rifampicin-resistant M. tuberculosis infection in clinical samples by line probe assay (INNO LiPA Rif-TB). New Microbiol 2004; 27: 221-7. [ Links ]
22. Cockerill FR III, Uhl JR, Temesgen Z, Zhang Y, Stockman L, Roberts GD, et al. Rapid identification of a point mutation of the Mycobacterium tuberculosis catalase-peroxidase (katG) gene associated with isoniazid resistance. J Infect Dis 1995; 171: 240-5. [ Links ]
23. Cole ST. Rifamycin resistance in mycobacteria. Res Microbiol 1996; 147: 48-52. [ Links ]
24. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998; 393: 537-44. [ Links ]
25. Coleri A, Cokmus C, Ozcan B, Akcelik M, Tukel C. Determination of antibiotic resistance and resistance plasmids of clinical Enterococcus species. J Gen Appl Microbiol 2004; 50: 213-9. [ Links ]
26. Corbett L, Raviglione M. Global burden of Tuberculosis: past, present and future. En: Cole ST, Jacobs WR, Davis Eisenach K, editors. Tuberculosis and the Tubercle Bacillus. Washington D.C., ASM Press, 2005, p. 3-12. [ Links ]
27. Cummings MP, Segal MR. Few amino acid positions in rpoB are associated with most of the rifampin resistance in Mycobacterium tuberculosis. BMC Bioinformatics 2004; 5: 137. [ Links ]
28. Davies AP, Billington OJ, Bannister BA, Weir WR, McHugh TD, Gillespie SH. Comparison of fitness of two isolates of Mycobacterium tuberculosis, one of which had developed multi-drug resistance during the course of treatment. J Infect 2000; 41: 184-7. [ Links ]
29. Davis IJ, Roberts AP, Ready D, Richards H, Wilson M, Mullany P. Linkage of a novel mercury resistance operon with streptomycin resistance on a conjugative plasmid in Enterococcus faecium. Plasmid 2005; 54: 26-38. [ Links ]
30. de Oliveira MM, da Silva RA, Cardoso OM, Gomes HM, Fonseca L, Werneck-Barreto AM, et al. Rapid detection of resistance against rifampicin in isolates of Mycobacterium tuberculosis from Brazilian patients using a reverse-phase hybridization assay. J Microbiol Methods 2003; 53: 335-42. [ Links ]
31. DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry III CE. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA 2000; 97: 9677-82. [ Links ]
32. Dessen A, Quemard A, Blanchard JS, Jacobs Jr WR, Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 1995; 267: 1638-41. [ Links ]
33. Dobner P, Rusch-Gerdes S, Bretzel G, Feldmann K, Rifai M, Loscher TR, et al. Usefulness of Mycobacterium tuberculosis genomic mutations in the genes katG and inhA for the prediction of isoniazid resistance. Int J Tuberc Lung Dis 1997; 1: 365-9. [ Links ]
34. Dye C, Espinal MA, Watt CJ, Mbiaga C, Williams BG. Worldwide incidence of multidrug-resistant tuberculosis. J Infect Dis 2002; 185: 1197-202. [ Links ]
35. Esiobu N, Armenta L, Ike J. Antibiotic resistance in soil and water environments. Int J Environ Health Res 2002; 12: 133-44. [ Links ]
36. Espinal MA, Dye C, Raviglione M, Kochi A. Rational ‘DOTS plus’ for the control of MDR-TB. Int J Tuberc Lung Dis 1999; 3: 561-3. [ Links ]
37. Espinal MA, Laserson K, Camacho M, Fusheng Z, Kim SJ, Tlali RE, et al. Determinants of drug-resistant tuberculosis: analysis of 11 countries. Int J Tuberc Lung Dis 2001; 5: 887-93. [ Links ]
38. Ezekiel DH, Hutchins JE. Mutations affecting RNA polymerase associated with rifampicin resistance in Escherichia coli. Nature 1968; 220: 276-7. [ Links ]
39. Ferrazoli L, Palaci M, Telles MA, Ueki SY, Kritski A, Marques LR, et al. Catalase expression, katG, and MIC of isoniazid for Mycobacterium tuberculosis isolates from Sao Paulo, Brazil. J Infect Dis 1995; 171: 237-40. [ Links ]
40. Fischer M. Investigation of a broad-spectrum PCR assay for human papillomaviruses in screening benign lesions of the upper aerodigestive tract. ORL J Otorhinolaryngol Relat Spec 2005; 67: 237-41. [ Links ]
41. Garcia de Viedma D, Lasala IF, Chaves F, Alcala L, Bouza E. New real-time PCR able to detect in a single tube multiple rifampin resistance mutations and high-level isoniazid resistance mutations in Mycobacterium tuberculosis. J Clin Microbiol 2002; 40: 988-95. [ Links ]
42. Garcia L, Alonso-Sanz M, Rebollo MJ, Tercero JC, Chaves F. Mutations in the rpoB gene of rifampin-resistant Mycobacterium tuberculosis isolates in Spain and their rapid detection by PCR-enzyme-linked immunosorbent assay. J Clin Microbiol 2001; 39: 1813-8. [ Links ]
43. Gfeller KY, Roth M, Meile L, Teuber M. Sequence and genetic organization of the 19.3-kb erythromycin- and dalfopristin-resistance plasmid pLME300 from Lactobacillus fermentum ROT1. Plasmid 2003; 50: 190-201. [ Links ]
44. Gonzalez N, Torres MJ, Palomares JC, Aznar J. Characterization of the rpoB gene mutations in clinical isolates of rifampicin-resistant Mycobacterium tuberculosis. Enferm Infecc Microbiol Clin 1998; 16: 404-7. [ Links ]
45. Goto M, Oka S, Tachikawa N, Kitada K, Wada M, Abe C, et al. KatG sequence deletion is not the major cause of isoniazid resistance in Japanese and Yemeni Mycobacterium tuberculosis isolates. Mol Cell Probes 1995; 9:433-439. [ Links ]
46. Hacker J, Carniel E. Ecological fitness, genomic islands and bacterial pathogenicity. A Darwinian view of the evolution of microbes. EMBO Rep 2001; 2: 376-81. [ Links ]
47. Haegi V. New aspects of tuberculosis therapy. Schweiz Med Wochenschr 1975; 105: 245-50. [ Links ]
48. Heifets LB. Antimycobacterial drugs. Semin Respir Infect 1994; 9: 84-103. [ Links ]
49. Heym B, Alzari PM, Honore N, Cole ST. Missense mutations in the catalase-peroxidase gene, katG, are associated with isoniazid resistance in Mycobacterium tuberculosis. Mol Microbiol 1995; 15: 235-45. [ Links ]
50. Heym B, Honore N, Truffot-Pernot C, Banerjee A, Schurra C, Jacobs Jr WR, et al. Implications of multidrug resistance for the future of short-course chemotherapy of tuberculosis: a molecular study. Lancet 1994; 344: 293-8. [ Links ]
51. Heym B, Saint-Joanis B, Cole ST. The molecular basis of isoniazid resistance in Mycobacterium tuberculosis. Tuber Lung Dis 1999; 79: 267-71. [ Links ]
52. Hirano K, Abe C, Takahashi M. Mutations in the rpoB gene of rifampin-resistant Mycobacterium tuberculosis strains isolated mostly in Asian countries and their rapid detection by line probe assay. J Clin Microbiol 1999; 37: 2663-6. [ Links ]
53. Hofling CC, Pavan EM, Giampaglia CM, Ferrazoli L, Aily DC, de Albuquerque DM, et al. Prevalence of katG Ser315 substitution and rpoB mutations in isoniazid-resistant Mycobacterium tuberculosis isolates from Brazil. Int J Tuberc Lung Dis 2005; 9: 87-93. [ Links ]
54. Jaber M, Rattan A, Kumar R. Presence of katG gene in resistant Mycobacterium tuberculosis. J Clin Pathol 1996; 49: 945-7. [ Links ]
55. Jacobs Jr WR, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, et al. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 1993; 260: 819-22. [ Links ]
56. Jarlier V, Nikaido H. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett 1994; 123: 11-8. [ Links ]
57. Jayakumar A, Tai MH, Huang WY, Al-Feel W, Hsu M, bu-Elheiga L, et al. Human fatty acid synthase: properties and molecular cloning. Proc Natl Acad Sci USA 1995; 92: 8695-9. [ Links ]
58. Jones LA, McIver CJ, Kim MJ, Rawlinson WD, White PA. The aadB gene cassette is associated with blaSHV genes in Klebsiella species producing extended-spectrum beta-lactamases. Antimicrob Agents Chemother 2005; 49: 794-7. [ Links ]
59. Kapur V, Li LL, Iordanescu S, Hamrick MR, Wanger A, Kreiswirth BN, et al. Characterization by automated DNA sequencing of mutations in the gene (rpoB) encoding the RNA polymerase beta subunit in rifampin-resistant Mycobacterium tuberculosis strains from New York City and Texas. J Clin Microbiol 1994; 32: 1095-8. [ Links ]
60. Khan AA, Srivastava R, Sinha VB, Srivastava BS. Regulation of toxin biosynthesis by plasmids in Vibrio cholerae. J Gen Microbiol 1985; 131: 2653-7. [ Links ]
61. Kiepiela P, Bishop KS, Smith AN, Roux L, York DF. Genomic mutations in the katG, inhA and aphC genes are useful for the prediction of isoniazid resistance in Mycobacterium tuberculosis isolates from Kwazulu Natal, South Africa. Tuber Lung Dis 2000; 80: 47-56. [ Links ]
62. Kugelberg E, Lofmark S, Wretlind B, Andersson DI. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J Antimicrob Chemother 2005; 55: 22-30. [ Links ]
63. Kuziora MA, Chalmers Jr JH, Douglas MG, Hitzeman RA, Mattick JS, Wakil SJ. Molecular cloning of fatty acid synthetase genes from Saccharomyces cerevisiae. J Biol Chem 1983; 258: 11648-53. [ Links ]
64. Larsen MH, Vilcheze C, Kremer L, Besra GS, Parsons L, Salfinger M, et al. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol Microbiol 2002; 46: 453-66. [ Links ]
65. Lavender C, Globan M, Sievers A, Billman-Jacobe H, Fyfe J. Molecular characterization of isoniazid-resistant Mycobacterium tuberculosis isolates collected in Australia. Antimicrob Agents Chemother 2005; 49: 4068-74. [ Links ]
66. Lee AS, Lim IH, Tang LL, Telenti A, Wong SY. Contribution of kasA analysis to detection of isoniazid-resistant Mycobacterium tuberculosis in Singapore. Antimicrob Agents Chemother 1999; 43: 2087-9. [ Links ]
67. Lee AS, Lim IH, Tang LL, Wong SY. High frequency of mutations in the rpoB gene in rifampin-resistant clinical isolates of Mycobacterium tuberculosis from Singapore. J Clin Microbiol 2005; 43: 2026-7. [ Links ]
68. Lee AS, Teo AS, Wong SY. Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 2001; 45: 2157-9. [ Links ]
69. Lee H, Cho SN, Bang HE, Lee JH, Bai GH, Kim SJ, et al. Exclusive mutations related to isoniazid and ethionamide resistance among Mycobacterium tuberculosis isolates from Korea. Int J Tuberc Lung Dis 2000; 4: 441-7. [ Links ]
70. Lehoux DE, Sanschagrin F, Kukavica-Ibrulj I, Potvin E, Levesque RC. Identification of novel pathogenicity genes by PCR signature-tagged mutagenesis and related technologies. Methods Mol Biol 2004; 266: 289-304. [ Links ]
71. Li XZ, Zhang L, Nikaido H. Efflux pump-mediated intrinsic drug resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 2004; 48: 2415-23. [ Links ]
72. Lipsitch M, Levin BR. Population dynamics of tuberculosis treatment: mathematical models of the roles of non-compliance and bacterial heterogeneity in the evolution of drug resistance. Int J Tuberc Lung Dis 1998; 2: 187-99. [ Links ]
73. Liu J, Barry III CE, Besra GS, Nikaido H. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J Biol Chem 1996; 1: 29545-51. [ Links ]
74. Macq JC, Theobald S, Dick J, Dembele M. An exploration of the concept of directly observed treatment (DOT) for tuberculosis patients: from a uniform to a customised approach. Int J Tuberc Lung Dis 2003; 7: 103-9. [ Links ]
75. Magnuson K, Jackowski S, Rock CO, Cronan Jr JE. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev 1993; 57: 522-42. [ Links ]
76. Mani C, Selvakumar N, Kumar V, Narayanan S, Narayanan PR. Comparison of DNA sequencing, PCR-SSCP and PhaB assays with indirect sensitivity testing for detection of rifampicin resistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 2003; 7: 652-9. [ Links ]
77. Mariam DH, Mengistu Y, Hoffner SE, Andersson DI. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2004; 48: 1289-94. [ Links ]
78. Marttila HJ, Soini H, Eerola E, Vyshnevskaya E, Vyshnevskiy BI, Otten TF, et al. A Ser315Thr substitution in KatG is predominant in genetically heterogeneous multidrug-resistant Mycobacterium tuberculosis isolates originating from the St. Petersburg area in Russia. Antimicrob Agents Chemother 1998; 42: 2443-5. [ Links ]
79. Mayta H, Gilman RH, Arenas F, Valencia T, Caviedes L, Montenegro SH, et al. Evaluation of a PCR-based universal heteroduplex generator assay as a tool for rapid detection of multidrug-resistant Mycobacterium tuberculosis in Peru. J Clin Microbiol 2003; 41: 5774-7. [ Links ]
80. McCammon MT, Gillette JS, Thomas DP, Ramaswamy SV, Graviss EA, Kreiswirth BN, et al. Detection of rpoB mutations associated with rifampin resistance in Mycobacterium tuberculosis using denaturing gradient gel electrophoresis. Antimicrob Agents Chemother 2005; 49: 2200-9. [ Links ]
81. Miesel L, Weisbrod TR, Marcinkeviciene JA, Bittman R, Jacobs Jr WR. NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J Bacteriol 1998; 180: 2459-67. [ Links ]
82. Mitchison DA. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 1985; 66: 219-25. [ Links ]
83. Mitchison D A. Basic mechanisms of chemotherapy. Chest 1979; 76: 771-81. [ Links ]
84. Mitchison DA, Nunn AJ. Influence of initial drug resistance on the response to short-course chemotherapy of pulmonary tuberculosis. Am Rev Respir Dis 1986; 133: 423-30. [ Links ]
85. Mokrousov I, Otten T, Vyshnevskiy B, Narvskaya O. Allele-specific rpoB PCR assays for detection of rifampin-resistant Mycobacterium tuberculosis in sputum smears. Antimicrob Agents Chemother 2003; 47: 2231-5. [ Links ]
86. Morlock GP, Metchock B, Sikes D, Crawford JT, Cooksey RC. EthA, inhA, and katG loci of ethionamide-resistant clinical Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 2003; 47: 3799-805. [ Links ]
87. Murinda SE, Liu SM, Roberts RF, Wilson RA. Colicinogeny among Escherichia coli serotypes, including O157:H7, representing four closely related diarrheagenic clones. J Food Prot 1998; 61: 1431-8. [ Links ]
88. Musser JM. Antimicrobial agent resistance in mycobacteria: molecular genetic insights. Clin Microbiol Rev 1995; 8: 496-514. [ Links ]
89. Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Embden JD. Characterization of the catalase-peroxidase gene (katG) and inhA locus in isoniazid-resistant and -susceptible strains of Mycobacterium tuberculosis by automated DNA sequencing: restricted array of mutations associated with drug resistance. J Infect Dis 1996; 173: 196-202. [ Links ]
90. Nikaido H, Jarlier V. Permeability of the mycobacterial cell wall. Res Microbiol 1991; 142: 437-43. [ Links ]
91. Noble WC. Antibiotic resistance in the staphylococci. Sci Prog 1997; 80 (Pt 1): 5-20. [ Links ]
92. Odriozola JM, Ramos JA, Bloch K. Fatty acid synthetase activity in Mycobacterium smegmatis. Characterization of the acyl carrier protein-dependent elongating system. Biochim Biophys Acta 1977; 488: 207-17. [ Links ]
93. Pablos-Mendez A, Knirsch CA, Barr RG, Lerner BH, Frieden TR. Nonadherence in tuberculosis treatment: predictors and consequences in New York City. Am J Med 1997; 102: 164-70. [ Links ]
94. Park YK, Kim BJ, Ryu S, Kook YH, Choe YK, Bai GH, et al. Cross-resistance between rifampicin and KRM-1648 is associated with specific rpoB alleles in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 2002; 6: 166-70. [ Links ]
95. Patterson JE. Extended-spectrum beta-lactamases. Semin Respir Crit Care Med 2003; 24: 79-88. [ Links ]
96. Peterson DO, Bloch K. Mycobacterium smegmatis fatty acid synthetase. Long chain transacylase chain length specificity. J Biol Chem 1977; 252: 5735-9. [ Links ]
97. Quemard A, Laneelle G, Lacave C. Mycolic acid synthesis: a target for ethionamide in mycobacteria. Antimicrob Agents Chemother 1992; 36: 1316-21. [ Links ]
98. Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs Jr WR, et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995; 34: 8235-41. [ Links ]
99. Ramaswamy S, Musser JM. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: Tuber Lung Dis 1998; 79: 3-29. [ Links ]
100. Raviglione MC, Pio A. Evolution of WHO policies for tuberculosis control, 1948-2001. Lancet 2002; 359: 775-80. [ Links ]
101. Raze D, Dardenne O, Hallut S, Martinez-Bueno M, Coyette J. Ghuysen JM. The gene encoding the low-affinity penicillin-binding protein 3r in Enterococcus hirae S185R is borne on a plasmid carrying other antibiotic resistance determinants. Antimicrob Agents Chemother 1998; 42: 534-9. [ Links ]
102. Riccio ML, Pallecchi L, Fontana R, Rossolini GM. In70 of plasmid pAX22, a bla(VIM-1)-containing integron carrying a new aminoglycoside phosphotransferase gene cassette. Antimicrob Agents Chemother 2001; 45: 1249-53. [ Links ]
103. Rindi L, Bianchi L, Tortoli E, Lari N, Bonanni D, Garzelli C. A real-time PCR assay for detection of isoniazid resistance in Mycobacterium tuberculosis clinical isolates. J Microbiol Methods 2003; 55: 797-800. [ Links ]
104. Riska PF, Su Y, Bardarov S, Freundlich L, Sarkis G, Hatfull G, et al. Rapid film-based determination of antibiotic susceptibilities of Mycobacterium tuberculosis strains by using a luciferase reporter phage and the Bronx Box. J Clin Microbiol 1999; 37: 1144-9. [ Links ]
105. Ristow M, Mohlig M, Rifai M, Schatz H, Feldmann K, Pfeiffer A. New isoniazid/ethionamide resistance gene mutation and screening for multidrug-resistant Mycobacterium tuberculosis strains. Lancet 1995; 346: 502-3. [ Links ]
106. Rock CO, Cronan JE. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim Biophys Acta 1996; 1302: 1-16. [ Links ]
107. Rojas L, Vinuesa T, Tubau F, Truchero C, Benz R, Vinas M. Integron presence in a multiresistant Morganella morganii isolate. Int J Antimicrob Agents 2006; 27: 505-12. [ Links ]
108. Rouse DA, Li Z, Bai GH, Morris SL. Characterization of the katG and inhA genes of isoniazid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 1995; 39: 2472-77. [ Links ]
109. Rouse DA, Morris SL. Molecular mechanisms of isoniazid resistance in Mycobacterium tuberculosis and Mycobacterium bovis. Infect Immun 1995; 63:1427-33. [ Links ]
110. Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, et al. Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother 2002; 46: 1204-11. [ Links ]
111. Schilke K, Weyer K, Bretzel G, Amthor B, Brandt J, Sticht-Groh V, et al. Universal pattern of RpoB gene mutations among multidrug-resistant isolates of Mycobacterium tuberculosis complex from Africa. Int J Tuberc Lung Dis 1999; 3: 620-6. [ Links ]
112. Severin A, Tabei K, Tenover F, Chung M, Clarke N. Tomasz. A. High level oxacillin and vancomycin resistance and altered cell wall composition in Staphylococcus aureus carrying the staphylococcal mecA and the enterococcal vanA gene complex. J Biol Chem 2004, 279: 3398-407. [ Links ]
113. Siddiqi N, Das R, Pathak N, Banerjee S, Ahmed N, Katoch VM, et al. Mycobacterium tuberculosis isolate with a distinct genomic identity overexpresses a tap-like efflux pump. Infection 2004; 32: 109-11. [ Links ]
114. Singh N, Wakil SJ, Stoops JK. Yeast fatty acid synthase: structure to function relationship. Biochemistry 1985; 24: 6598-602. [ Links ]
115. Sintchenko V, Chew WK, Jelfs PJ, Gilbert GL. Mutations in rpoB gene and rifabutin susceptibility of multidrug-resistant Mycobacterium tuberculosis strains isolated in Australia. Pathology 1999; 31: 257-60. [ Links ]
116. Sintchenko V, Jelfs PJ, Chew WK, Gilbert GL. Mycobacterium tuberculosis rpoB gene DNA sequencing: implications for detection of rifamycin resistance. J Antimicrob Che-mother 1999; 44: 294-5. [ Links ]
117. Smith MA, Bidochka MJ. Bacterial fitness and plasmid loss: the importance of culture conditions and plasmid size. Can J Microbiol 1998; 44: 351-5. [ Links ]
118. Spindola de Miranda MS, Kritski A, Filliol I, Mabilat C, Panteix G, Drouet E. Mutations in the rpoB gene of rifam-picin-resistant Mycobacterium tuberculosis strains isolated in Brazil and France. Mem Inst Oswaldo Cruz 2001; 96: 247-50. [ Links ]
119. Strizhkov BN, Drobyshev AL, Mikhailovich VM, Mirzabekov AD. PCR amplification on a microarray of gel-immobilized oligonucleotides: detection of bacterial toxin -and drug-resistant genes and their mutations. Biotechniques 2000; 29: 844-54. [ Links ]
120. Takayama K. Selective action of isoniazid on the synthesis of cell wall mycolates in mycobacteria. Ann N Y Acad Sci 1974; 235: 426-38. [ Links ]
121. Takayama K, Armstrong EL, David HL. Restoration of mycolate synthetase activity in Mycobacterium tuberculosis exposed to isoniazid. Am Rev Respir Dis 1974; 110: 43-8. [ Links ]
122. Takayama K, Datta AK. Structure-to-function relationship of mycobacterial cell envelope components. Res Microbiol 1991; 142: 443-8. [ Links ]
123. Takayama K, Schnoes HK, Armstrong EL, Boyle RW. Site of inhibitory action of isoniazid in the synthesis of mycolic acids in Mycobacterium tuberculosis. J Lipid Res 1975; 16: 308-17. [ Links ]
124. Takayama K, Wang L, David HL. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother 1972; 2: 29-35. [ Links ]
125. Telenti A, Honore N, Bernasconi C, March J, Ortega A, Heym B, et al. Genotypic assessment of isoniazid and rifampin resistance in Mycobacterium tuberculosis: a blind study at reference laboratory level. J Clin Microbiol 1997; 35: 719-23. [ Links ]
126. Telenti A, Imboden P, Marchesi F, Lowrie D, Cole S, Colston MJ, et al. Detection of rifampicin-resistance mutations in Mycobacterium tuberculosis. Lancet 1993; 341: 647-50. [ Links ]
127. Torres MJ, Criado A, Palomares JC, Aznar J. Use of real-time PCR and fluorimetry for rapid detection of rifampin and isoniazid resistance-associated mutations in Mycobacterium tuberculosis. J Clin Microbiol 2000; 38: 3194-9. [ Links ]
128. Torres MJ, Criado A, Ruiz M, Llanos AC, Palomares JC, Aznar J. Improved real-time PCR for rapid detection of rifampin and isoniazid resistance in Mycobacterium tuberculosis clinical isolates. Diagn Microbiol Infect Dis 2003; 45: 207-12. [ Links ]
129. Valway SE, Sanchez MP, Shinnick TF, Orme I, Agerton T, Hoy D, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N Engl J Med 1998; 338: 633-9. [ Links ]
130. van Doorn HR, Claas EC, Templeton KE, Van Der Zanden AG, te Kopele Vije A, de Jong MD, et al. Detection of a point mutation associated with high-level isoniazid resistance in Mycobacterium tuberculosis by using real-time PCR technology with 3'-minor groove binder-DNA probes. J Clin Microbiol 2003; 41: 4630-5. [ Links ]
131. Vannelli TA, Dykman A, Ortiz de Montellano PR. The anti-tuberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem 2002; 277: 12824-9. [ Links ]
132. Viader-Salvado JM, Luna-Aguirre CM, Reyes-Ruiz JM, Valdez-Leal R, del Bosque-Moncayo ML, Tijerina-Menchaca R, et al. Frequency of mutations in rpoB and codons 315 and 463 of katG in rifampin- and/or isoniazid-resistant Mycobacterium tuberculosis isolates from northeast Mexico. Microb Drug Resist 2003; 9: 33-8. [ Links ]
133. Vilcheze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, et al. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J Bacteriol 2000; 182: 4059-67. [ Links ]
134. Vilcheze C, Weisbrod TR, Chen B, Kremer L, Hazbon MH, Wang F, et al. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother 2005; 49: 708-20. [ Links ]
135. Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989; 28: 4523-30. [ Links ]
136. Wang L, Takayama K. Relationship between the uptake of isoniazid and its action on in vivo mycolic acid synthesis in Mycobacterium tuberculosis. Antimicrob Agents Chemother 1972; 2: 438-41. [ Links ]
137. Watterson SA, Wilson SM, Yates MD, Drobniewski FA. Comparison of three molecular assays for rapid detection of rifampin resistance in Mycobacterium tuberculosis. J Clin Microbiol 1998; 36: 1969-73. [ Links ]
138. Wayne LG. Dormancy of Mycobacterium tuberculosis and latency of disease. Eur J Clin Microbiol Infect Dis 1994; 13: 908-14. [ Links ]
139. Winder F. Catalase and peroxidase in mycobacteria. Possible relationship to the mode of action of isoniazid. Am Rev Respir Dis 1960; 81: 68-78. [ Links ]
140. Winder F, Brennan P. The accumulation of free trehalose by mycobacteria exposed to isoniazid. Biochim Biophys Acta 1964; 90: 442-4. [ Links ]
141. Winder F, Collins P. The effect of isoniazid on nicotinamide nucleotide levels in Mycobacterium bovis, strain BCG. Am Rev Respir Dis 1968; 97: 719-20. [ Links ]
142. Winder FG, Brennan P, Ratledge C. Synthesis of fatty acids by extracts of mycobacteria and the absence of inhibition by isoniazid. Biochem J 1964; 93: 635-40. [ Links ]
143. Winder FG, Brennan PJ, McDonnell I. Effects of isoniazid on the composition of mycobacteria, with particular reference to soluble carbohydrates and related substances. Biochem J 1967; 104: 385-93. [ Links ]
144. Winder FG, Collins P. The effect of isoniazid on nicotinamide nucleotide concentrations in tubercle bacilli. A correction and further observations. Am Rev Respir Dis 1969; 100: 101-3. [ Links ]
145. Winder FG, Collins P, Rooney SA. Effects of isoniazid on mycolic acid synthesis in Mycobacterium tuberculosis and on its cell envelope. Biochem J 1970; 117: 27P. [ Links ]
146. Winder FG, Collins PB. Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J Gen Microbiol 1970; 63: 41-8. [ Links ]
147. Winder FG, Rooney SA. The effects of isoniazid on the carbohydrates of Mycobacterium tuberculosis BCG. Biochem J 1970; 117: 355-68. [ Links ]
148. Winder FG, Rooney SA. Effects of isoniazid on the triglycerides of BCG. Am Rev Respir Dis 1968; 97: 938-40. [ Links ]
149. Winder FG, Rooney SA. The effect of isoniazid on the alkali-extractable polysaccharides of Mycobacterium tuberculosis. Biochem J 1968; 110: 8P-9P. [ Links ]
150. Yu S, Chouchane S, Magliozzo RS. Characterization of the W321F mutant of Mycobacterium tuberculosis catalase-peroxidase KatG. Protein Sci 2002; 11: 58-64. [ Links ]
151. Zhang Y. Genetic basis of isoniazid resistance of Mycobacterium tuberculosis. Res Microbiol 1993; 144: 143-9. [ Links ]
152. Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992; 358: 591-3. [ Links ]
153. Zhao JR, Bai YJ, Wang Y, Zhang QH, Luo M, Yan XJ. Development of a pyrosequencing approach for rapid screening of rifampin, isoniazid and ethambutol-resistant Mycobacterium tuberculosis. Int J Tuberc Lung Dis 2005; 9: 328-32. [ Links ]
Recibido: 31/10/05
Aceptado: 5/6/06

