










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista argentina de microbiología
Print version ISSN 0325-7541On-line version ISSN 1851-7617
Rev. argent. microbiol. vol.41 no.3 Ciudad Autónoma de Buenos Aires July/Sept. 2009
Infección por el virus de la Lengua azul: activación de señales celulares que inducen apoptosis
E. Mortola*, A. Larsen
Inmunología Veterinaria, Facultad de Ciencias Veterinarias, Universidad Nacional de La Plata, 60 y 118, (1900) La Plata, Argentina
*Correspondencia. E-mail: mortola@fcv.unlp.edu.ar
RESUMEN
El virus de la Lengua azul (VLA) es un ARN virus de doble cadena que induce apoptosis tanto en cultivos celulares como en tejidos blanco. Con el fin de dilucidar el mecanismo de apoptosis en la infección por el VLA, en el presente trabajo examinamos en detalle, por la técnica de Western blot, las señales celulares de caspasas, Bax, citocromo c, Smac/DIABLO y factor nuclear kappa B (NF-kB) que se activan en la infección viral. Hemos comprobado que luego de la infección in vitro con el VLA, se detectó la activación de la caspasa 8 y con ello el mecanismo extrínseco de la apoptosis. También detectamos por primera vez no sólo la activación de miembros de la familia Bcl-2 (Bax), sino también la liberación del citocromo c y la proteína Smac/DIABLO, confirmando que en la infección por el VLA está involucrado el mecanismo secuencial intrínseco de la apoptosis. Asimismo, demostramos que la infección por el VLA activa el NF-kB y que la apoptosis es sustancialmente reducida mediante la inhibición del mismo. La activación de las señales celulares tales como Bax, citocromo c, Smac/DIABLO y NF-kB presentados en este trabajo, esclarecen los mecanismos apoptóticos durante la infección por el VLA para una mayor comprensión del papel primario que juega la apoptosis en la patogénesis del virus.
Palabras clave: Apoptosis; Virus de la Lengua azul; Bax; Citocromo c; Smac/DIABLO; NF-kB
ABSTRACT
Bluetongue virus infection: signaling pathway activated during apoptosis. Bluetongue (BTV) is a double-stranded RNA virus that induces apoptosis both in mammalian cell cultures and in target tissues. To elucidate the apoptosis pathways in BTV infection, we have examined in detail the apoptosis mechanism by examination of caspases, Bax, cytochrome c, Smac/DIABLO and NF-kB signalling pathways. In this report, after cell infection with BTV, the activation of caspase 8 was detected, proving the extrinsic receptor binding apoptotic pathway. Apoptosis followed a sequential pathway involving the detection of activated Bcl-2 family members. Furthermore, its translocation to the mitochondria, as well as the release of cytochrome c and Smac/Diablo confirmed that BTV apoptosis involves the sequential intrinsic pathway. In addition, we demonstrated that NF-kB was activated following BTV infection and cell treatment with an inhibitor peptide before BTV infection, prevented NF-kB activation and substantially reduced cellular apoptosis. Our accumulating data concerning the activation of Bax, cytochrome c, Smac/DIABLO and NF-kB clarify the mechanism of apoptosis during BTV infection, and confer a better understanding of the primary role of apoptosis in BTV pathogenesis.
Key words: Apoptosis; Bluetongue virus; Bax; Cytochrome c; Smac/DIABLO; NF-kB
INTRODUCCIÓN
El virus de la Lengua azul (VLA) es un virus ARN de doble cadena que pertenece a la familia Reoviridae. La infección por la mayoría de los reovirus causa un efecto citopático (ECP) en la célula blanco y bajo ciertas condiciones algunos de estos virus han demostrado inducir apoptosis celular (7). El VLA induce apoptosis en cultivos celulares y en tejidos blanco in vivo, demostrando que este mecanismo de muerte celular juega un papel preponderante en la patofisiología de la infección. (11, 12, 26).
En trabajos previos hemos demostrado que la inducción de apoptosis por el VLA se debe a la acción de las proteínas de la cápside externa del virus (26). El tratamiento de cultivos celulares con una combinación de la proteína recombínante VP2 responsable de la unión al receptor de membrana y la proteína recombinante VP5 que actúa en la penetración de la membrana celular, fue suficiente para disparar apoptosis en la célula blanco. Hemos establecido asimismo que la inducción de apoptosis por la activación de la caspasa 3, también llamada caspasa ejecutora, está involucrada en el evento. Del mismo modo, se ha reportado la activación de las caspasas 8 y 9 en la infección por el VLA (27). Sin embargo, en la actualidad los mecanismos que actúan en la inducción de apoptosis por el VLA no están completamente dilucidados y son en gran parte revelados con las experiencias realizadas en el presente trabajo.
De acuerdo con el esquema convencional, hay por lo menos dos amplias vías que conducen a la apoptosis, una vía extrínseca y otra intrínseca. La vía extrínseca está mediada por ligandos de muerte (8, 9) que transmiten una señal al interior de la célula con la activación de la caspasa 8 iniciadora y subsecuentemente, la activación de la caspasa 3 efectora que desencadena la apoptosis (9, 18). En la vía intrínseca, la apoptosis se dispara por señales internas que llevan a la permea-bilización de la membrana mitocondrial, que causa la liberación del citocromo c del interior de la mitocondria al citosol, el cual desencadena la activación de la caspasa 9. Otra proteína liberada por el daño mitocondrial es el Smac/DIABLO. Este factor contrarresta el efecto de los factores llamados IAPs que son inhibidores de la apoptosis, los cuales normalmente están unidos y previenen la activación de la caspasa 3. La interacción precisa y la evidencia de la activación entre todos estos componentes del mecanismo de apoptosis es fundamental para la vía intrínseca. Ambas vías se unen a nivel de la caspasa 3 y subsecuentemente resulta en un proceso de degradación del ADN y empaquetamiento de la célula en pequeñas porciones que son tomadas por las células vecinas (5, 34).
Es bien conocido que el factor nuclear kappa B (NF-kB) es central en la regulación de apoptosis. En la mayoría de los casos, el NF-kB actúa como un regulador antiapoptótico, inhibiendo la muerte celular y logrando su sobrevida (2). Sin embargo, en otros casos la activación del NF-kB puede deberse a factores internos o externos de estrés. En las células en reposo, el NF-kB existe como un heterodímero de dos subunidades p50 y p65 que es mantenido en estado inactivo en el citosol por la unión con su inhibidor específico. El NF-kB puede ser liberado y degradado, resultando su liberación y su trans-locación al núcleo donde cumple una función de regulador transcripcional de varios genes relacionados (4, 6).
Los estudios aquí realizados son la continuación de hallazgos previos sobre la inducción de apoptosis por las proteínas de la cápside externa del VLA en células mamíferas. Con las experiencias realizadas en este trabajo, demostramos que seguido a la infección por el VLA, los eventos apoptóticos en células HeLa no sólo disparan la vía extrínseca sino también se demostró la activación de las señales celulares de Bax, citocromo c y Smac/DIABLO que prueban la vía mitocondrial de apoptosis. Por primera vez se estableció que este proceso fue acompañado por la activación del NF-kB, el cual parece jugar un papel importante en la inducción de apoptosis por el VLA. Los datos aquí presentados esclarecen el fenómeno de apoptosis tisular que ocurre luego de la infección viral, el cual tendría un papel primario en la patogénesis del VLA.
MATERIALES Y MÉTODOS
Cultivo celular, infección viral y reactivos utilizados
Las células epiteliales de carcinoma cervical humano (HeLa) se mantuvieron en medio Eagle modificado por Dulbecco (Gibco BRL, Gaithersburg, MD), conteniendo 10% de suero fetal bovino (SFB), 100 U/ml de penicilina y 100 g/ml de estreptomicina (Sigma-Aldrich Chemical Co., St. Louis, Mo) e incubaron a 37 °C con 5% de CO2.
El VLA serotipo 10 (VLA-10) titulado por ensayo de placa a 2,5 x 107 UFP/ml se utilizó para la infección de los cultivos celulares. Las células en monocapa se lavaron con medio de cultivo libre de SFB y fueron incubadas con el virus a una multiplicidad de infección (MOI) de 20 PFU/célula en medio de cultivo libre de SFB.
Los anticuerpos policlonales anti-caspasa 8, anti-caspasa 9 y anti-NF-kB (p50)/NF-kB (p65) se obtuvieron de Oncogene, CNbiosciences, Inc. Darmstadt, Alemania. Los anticuerpos monoclonales anti-caspasa 3 se obtuvieron de eBioscience, San Diego, CA. y los anticuerpos policlonales anti-Bax, anti-Citocromo c, anti-Smac/DIABLO y el péptido inhibidor del NF-kB (SN50) se obtuvieron de Calbiochem, Merck Biosciences, Darmstadt, Alemania.
Preparación de los lisados celulares para la detección de las caspasas y el NF-kB
Para detectar la actividad de las caspasas, aproximadamente 106 células fueron recolectadas y lavadas dos veces con una solución de fosfatos salina buferada (PBS), luego resuspendidas y lisadas en 50 mM PIPES/KOH-pH 6,5; 2 mM EDTA; 0,1% CHAPS; 5 mM dithiothreitol (DTT); cocktail inhibidor de proteasas y 10 µM fenil-metil-sulfonil-fluoruro (PMSF). Las células fueron centrifugadas a 200 g por 10 min a 4 °C y sometidas a ciclos de congelación y descongelación en hielo seco y metanol. El lisado celular fue centrifugado a 20.000 g a 4 °C por 30 min y se recuperó el sobrenadante. Los extractos celulares se analizaron por la técnica de Western blot.
Para la detección del NF-kB (p50 y p65) en el extracto nuclear de la célula, aproximadamente 106 células fueron lavadas con PBS frío e incubadas a 4 °C por 20 min en buffer (10 mM HEPES-pH 7,9; 10 mM KCl; 1,5 mM MgCl2; 0,5 mM DTT; cocktail inhibidor de proteasas y 0,5 µM PMSF). A cada muestra se le adicionó Nonidet P-40 a 0,5% (peso/volumen) de concentración final, seguido de una centrifugación a 10.000 g por 15 min. Cada pellet fue resuspendido y mantenido en buffer (20 mM HEPES- pH 7,9; 0,5 M NaCl; 1,5 mM MgCl2; 0,2 mM EDTA; 25% glicerol; 0,5 mM DTT; cocktail inhibidor de proteasas y 0,5 µM PMSF) a 4 °C por 3 h. Las muestras fueron centrifugadas nuevamente de igual forma y el sobrenadante se utilizó para la detección del NF-kB por la técnica de Western blot.
Extracción de las fracciones mitocondriales y citosólicas
Aproximadamente 106 células fueron lavadas dos veces con PBS helado, centrifugadas a 200 g por 10 min a 4 °C en tubo previamente enfriado, y el sedimento resuspendido en 250 µl de buffer helado (200 mM manitol; 20 mM HEPES (pH 7,4); 10 mM KCl; 1,5 mM MgCl2; 1 mM EDTA y 1 mM EGTA). Inmediatamente antes de usar, el buffer de extracción fue suplementado con cocktail inhibidor de proteasas, 10 µM PMSF y 1 mM DTT. Las células fueron mantenidas en hielo por 30 min, homogeneizadas cuidadosamente con un homogeneizador Dounce (entre 5 a 10 movimientos) y el lisado celular fue analizado por la coloración de azul de Trypan. Las muestras homogeneizadas fueron centrifugadas a 750 g por 5 min a 4 °C y el sobrenadante sujeto a otra nueva centrifugación a 10.000 g por 10 min a 4 °C. Los sedimentos conteniendo el material mitocondrial se resuspendieron en buffer de muestra. El sobrenadante fue sometido a ultracentrifugación a 100.000 g por 45 min a 4 °C; esta porción contiene la fracción citosólica. Sólo las muestras que no mostraron contaminación cruzada entre material mitocondrial y citosólico fueron usadas para las determinaciones. Ambas fracciones fueron conservadas a -70 °C hasta el análisis pertinente.
Técnica de Western blot
Brevemente, luego de los procedimientos indicados precedentemente sobre el tratamiento de las células, las muestras fueron lisadas en igual volumen de buffer de muestra con SDS y 2,5% 2-ME. Las proteínas fueron desnaturalizadas y reducidas por calor mediante el calentamiento de las muestras a 95 °C por 5 min. Se sembraron 20 µl de muestra en cada pocillo que contenía entre 50 a 100 µg de proteína. Las muestras fueron sometidas a SDS-PAGE (Sodium Dodecyl Sulphate - PolyAcryla-mide Gel Electrophoresis) en geles de 6-12% de acuerdo al peso molecular de la proteína buscada, y transferida a una membrana de nitrocelulosa (Gibco BRL, Gaithersburg, MD) siguiendo el protocolo estándar de transferencia. La membrana se bloqueó por 1 h a temperatura ambiente con PBS conteniendo 0,1% de tween-20 y 5% de leche descremada. La incubación con el anticuerpo primario y el secundario fue específica para la proteína celular buscada, siguiendo los requerimientos y condiciones de las instrucciones de la compañía que proveyó cada anticuerpo. Las bandas inmunorreactivas fueron detectadas por procedimientos de quimioluminiscencia mejorada utilizando películas de quimioluminiscencia de alta calidad (Hyperfilm ECL, Amersham).
Cuantificación de apoptosis
Para cuantificar la apoptosis relativa se empleó un equipo de ELISA para nucleosomas, basado en un ensayo no isotópico para cuantificar in vitro nucleosomas libres en células apoptóticas. El análisis se realizó siguiendo las instrucciones de la compañía (Calbiochem, Merck Biosciences, Darmstadt, Alemania).
RESULTADOS
Activación de la cascada de caspasas
Para determinar cómo se inicia la vía intrínseca y/o la extrínseca de apoptosis en la infección con el VLA, examinamos la activación de las caspasas 8 y 9 por la técnica de Western blot, utilizando anticuerpos que reconocen específicamente niveles endógenos de caspasa 8 activada y la forma entera y el fragmento mayor de la caspasa 9 resultante del clivaje de la misma. En la Figura 1A, la activación de la caspasa 8 se visualiza por un marcado incremento de su subunidad de 18 kDa a las 12 y 18 h luego de la exposición del virus en las células HeLa (MOI de 20). Estos resultados sugieren que la vía del receptor de muerte celular de membrana podría estar involucrada en la inducción de apoptosis en células HeLa por el VLA.

Figura 1. Activación de la cascada de caspasas. Análisis por Western blot de células HeLa a las 12 y 18 h p.i. con el VLA a un MOI de 20, mostrando (A) la actividad de la caspasa 8 representada por la fracción de 18 kDa; (B) la actividad de la caspasa 9 representada por la fracción de 35 kDa, resultante del clivaje de la enzima total de 48 kDa y (C) la actividad de la caspasa 3 representada por la fracción de 17 kDa resultante del clivaje de la enzima total de 32 kDa.
En la cascada de activación de la apoptosis, la caspasa 9 es una pro enzima que puede ser clivada durante la vía intrínseca. En la Figura 1B se observa que el fragmento mayor de 35 kDa de la caspasa 9 resultante del clivaje de la enzima entera de 48 kDa, aumenta considerablemente a las 12 y 18 h post-infección (p.i) viral en células HeLa (MOI de 20), cuando se las compara con el control celular no infectado. Estos resultados demuestran que la caspasa 9 es activada durante la infección por el VLA. En el paso siguiente de la activación en cascada, las dos vías convergen y ambas caspasas 8 y 9 activan la caspasa 3 ejecutora, que requiere de un proceso proteolítico donde el clivaje de la proteína libera el fragmento de 17 kDa que indica su activación (Figura 1C). Tomados en conjunto, nuestros resultados sugieren que la infección por el VLA activa las caspasas 8, 9 y 3 en un estadio temprano de la infección.
La vía mitocondrial de la apoptosis: activación de Bax y su translocación a la mitocondria
Debido a que la familia de proteínas Bcl-2 juega un papel fundamental en la inducción de la apoptosis por una variedad de virus (13, 25), analizamos en el presente trabajo la expresión de la proteína Bax, un miembro de la familia Bcl-2 que promueve la apoptosis a través de la vía intrínseca. Las proteínas del tipo Bax comprometen a la función mitocondrial y básicamente median los efectos de diversos inductores de muerte celular que causan permeabilidad de la membrana mitocondrial externa (33). La Figura 2A surge del análisis por la técnica de Western blot de la proteína Bax, en el citosol y en la fracción mitocondrial del lisado de células HeLa infectadas por el VLA. Concordante con la detección de niveles de expresión de proteína Bax en el citosol de células control no infectadas, se encontró un incremento de la proteína Bax en la fracción mitocondrial de las células HeLa infectadas por el VLA (MOI de 20) visualizado por la detección de una banda de 21 kDa a las 12 h p.i. en comparación con el control de células no infectadas.

Figura 2. Señales celulares internas en la vía mitocondrial de la apoptosis. Análisis por Western blot de las fracciones mitocondrial (Mit) y citosólica (Cit) de células HeLa infectadas con el VLA a un MOI de 20, mostrando (A) un incremento significativo de la proteína Bax en la fracción Mit de células infectadas (12 h p.i.) representada por la fracción de 21 kDa, cuando se las compara con el control celular no infectado; la activación de (B) citocromo c y (C) Smac/DIABLO a las 12 y 18 h p.i. representado por la presencia de una banda de 12 kDa que representa la forma activa del citocromo c y de 25 kDa de Smac/DIABLO en la fracción Cit, comparado con el control celular no infectado. Los controles positivos se realizaron con lisados celulares totales.
La infección por el VLA causa la liberación del citocromo c y Smac/DIABLO de la mitocondria
El citocromo c fue identificado como el primer factor pro apoptótico que, liberado por la mitocondria al citosol, se asocia con el aumento de la permeabilidad de la membrana mitocondrial (14, 36). El Smac/DIABLO, proteína similar al citocromo c, es liberado de la mitocondria durante la apoptosis y su función principal es liberar de la inhibición impuesta por las proteínas inhibidoras de apoptosis (IAPs) sobre las caspasas 9, 3 y 7. En este experimento nosotros investigamos si la permeabilidad de la membrana mitocondrial está asociada con la liberación de citocromo c y la apoptosis luego de la infección por el VLA. Con este fin, a nivel celular examinamos por la técnica de Western blot, la presencia del citocromo c y Smac/DIABLO en la fracción citosólica de células HeLa infectadas con el VLA (MOI de 20). Las Figuras 2B y 2C muestran en la fracción citosólica de las células (12 y 18 h p.i.), la presencia de las bandas de 12 kDa y 25 kDa correspondientes a las formas activas de citocromo c y Smac/DIABLO, respectivamente. Este hallazgo prueba la liberación de esas proteínas de la mitocondria al citosol durante la apoptosis inducida por el VLA, cuando se las compara con el control no infectado, el cual corresponde a la fracción celular citosólica que sufrió el mismo tratamiento de la muestra pero sin la infección viral. Se realizaron controles positivos para detectar citocromo c y Smac/DIABLO con lisado celular total sin el fraccionamiento de la porción mitocondrial de la citosólica.
La inhibición del NF-kB reduce significativamente la inducción de apoptosis por el VLA
Para comprobar si la activación del NF-kB es esencial en el proceso de apoptosis por la infección por el VLA, investigamos el bloqueo del mismo en cultivos de células mamíferas. El análisis por Western blot y quimiolu-miniscencia para detectar el NF-kB en los extractos nucleares se realizó utilizando dos anticuerpos policlonales anti-p50 y anti-p65. Ambas subunidades p65 y p50 fueron detectadas en los extractos nucleares de las células a las 12 y 18 h p.i., demostrando la translocación del NF- kB al núcleo celular (Figura 3A). Con el fin de dilucidar si la activación del NF-kB es esencial en la inducción de apoptosis, se procedió al bloqueo del factor nuclear mediante el uso de SN50, un péptido inhibidor con permeabilidad celular. Las células HeLa fueron previamente tratadas con SN50 por 30 min y luego infectadas con VLA (MOI de 20). Como se muestra en la Figura 3B, luego de 12 y 18 h p.i., en las células tratadas con SN50 se detectó una inhibición de la translocación nuclear de las subunidades p50 y p65 del NF-kB. El efecto del SN50 alcanzó un máximo de inhibición a una concentración extracelular de 50 g/ml (18 µM). A esta concentración, SN50 bloqueó la activación del NF-kB a las 12 h p.i. y la cuantificación de apoptosis por ELISA reveló que la inducción de apoptosis por el VLA en células HeLa se redujo en aproximadamente un 75% comparado con el control celular no tratado (Figura 3C). Estos resultados en conjunto sugieren que el NF-kB juega un papel importante en el mecanismo de apoptosis por el VLA.
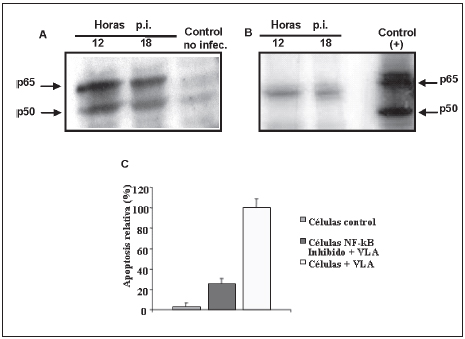
Figura 3. La vía de activación del NF-kB en la infección por el VLA. Análisis por Western blot de células HeLa a las 12 y 18 h p.i con el VLA a un MOI de 20, mostrando (A) la activación del NF-kB representada por la detección de las subunidades p65 y p50 cuando se las compara con los controles de células no infectadas y (B) el bloqueo de la activación del NF-kB por el uso del péptido inhibidor SN50 representado por la ausencia de las bandas correspondientes a las subunidades p50 y p65. Los controles positivos se realizaron con células no tratadas con el péptido inhibidor. (C) Cuantificación relativa de apoptosis por ELISA específica para nucleosomas libres, en células HeLa 12 h p.i. con el VLA (MOI de 20) o tratadas con el inhibidor del NF-kB. La apoptosis en células HeLa infectadas por el VLA (MOI de 20) fue establecida como el 100%. Los otros valores se expresaron en forma relativa al estándar, basados en los resultados de ELISA. Los valores corresponden a tres experimentos independientes con sus respectivos desvíos estándar.
DISCUSIÓN
Una gran variedad de virus tienen la capacidad de inducir apoptosis (30), y durante este proceso se activan una variedad de señales llevando a las células infectadas a una muerte por apoptosis. Tal es el caso de los reovirus y virus sindbis donde la apoptosis se induce tanto por la vía receptor celular como por la vía mitocondrial, ambas vías dependientes de las caspasas (10, 15, 18). En nuestros estudios previos, hemos demostrado que la inducción de apoptosis en células mamíferas por el VLA ocurre muy tempranamente luego de la infección y se debe a la internalización de la partícula viral y la subsecuente activación de la VP5 por la acidificación de las vesículas endosomales (26). Sin embargo, la vía celular por la cual el VLA induce apoptosis no está aún esclarecida. En el presente trabajo hemos realizado una serie de estudios para dilucidar las señales celulares que se activan en la inducción de apoptosis en células mamíferas infectadas por el VLA, basados en un ciclo de replicación viral tal como fue establecido en nuestros trabajos previos (26). Se utilizó un MOI de 20 (20 TCID50 por célula) para asegurar que el máximo número de células se infecten al inicio, y se revelaron importantes hallazgos en la inducción de apoptosis por el VLA. Primeramente, nuestros datos mostraron que el VLA induce apoptosis a través de las caspasas y que se activan ambas vías, extrínseca e intrínseca. En segundo término, que el NF-kB regula la inducción de apoptosis por el VLA.
Nuestros resultados ofrecen dos potenciales mecanismos de activación donde las caspasas son protagonistas. Uno es la activación de la caspasa 8 en la vía extrínseca de apoptosis causada por el VLA, comprobada por la identificación de la forma activa de la caspasa 8 por la técnica de Western blot. Seguido a esto, la caspasa 3 efectora es activada y el ADN fragmentado como consecuencia del proceso de apoptosis causado por el VLA. Los trabajos de Liu et al. (38) han demostrado que la caspasa 8 no sólo está involucrada en la activación extrínseca del mecanismo de apoptosis, sino también que la inhibición de la actividad de la caspasa 8 inhibe la liberación de citocromo c de la mitocondria, indicando la actividad de esta caspasa en el mecanismo intrínseco de apoptosis. En términos generales, la activación de la caspasa 8 es mediada por moléculas de la superficie celular como Fas o Fas ligando o receptor del factor de necrosis tumoral (TNFR). Este concepto se correlaciona con nuestros hallazgos previos donde el tratamiento de las células con proteínas recombinantes de la cápside externa del VLA, así como también la infección con el VLA inactivado por rayos UV, son suficientes para disparar apoptosis (26). Es muy posible que el VLA active la caspasa 8 durante la entrada a la célula. Estos resultados sugieren que la activación de la vía mitocondrial podría ser un evento secundario seguido a la señal de la caspasa 8. Por lo tanto, la apoptosis se inicia por la activación de un receptor de muerte pero su expresión completa requeriría de la participación de la vía mitocondrial (19).
Los resultados volcados en este manuscrito muestran que la inducción de apoptosis por el VLA está asociada con la translocación de la proteína Bax a la mitocondria. Nosotros hemos observado que el clivaje de Bax a las 12 h posteriores a la infección se realizó en conjunto con la temprana e importante activación de la caspasa 8. Igualmente, la proteína Bax activada se transloca a la mitocondria derivando en la pérdida del potencial de membrana e induciendo la liberación del citocromo c, mediando así la activación de la caspasa (39). El citocromo c causa una alteración en el poro de la membrana, que es un paso irreversible hacia la apoptosis (1, 29). La detección de la pérdida del potencial de membrana por sí solo no es suficiente para aseverar la participación de la vía intrínseca de la apoptosis, si no se la asocia a la detección de la activación de factores celulares involucrados en tal proceso (3, 34, 37). Nagaleekar et al. (27) establecen que una alteración en la permeabilidad de la membrana mitocondrial en la infección por el VLA es suficiente para afirmar la activación de la vía intrínseca de la apoptosis, sin establecer la activación de las señales celulares involucradas. Sin embargo, está comprobado que una pérdida en el potencial de membrana mitocondrial no es un requerimiento temprano y universal en la inducción de apoptosis, como fue concebido inicialmente (24). Una mayor y completa comprensión de la relación entre la pérdida del potencial de membrana propuesta por Nagaleekar et al. (27) y las señales celulares activadas que quedan demostradas en el presente trabajo, es requisito previo para que podamos precisar con exactitud la activación del mecanismo intrínseco de la apoptosis en la infección por el VLA.
En los experimentos desarrollados en este trabajo, el citocromo c fue detectado a las 12 h, en paralelo con la aparición de la forma activa de la caspasa 9. Estos datos confirman la idea de que la apoptosis inducida por el VLA se desarrolla a través de la mitocondria, dependiendo de la activación de las caspasas. Hemos demostrado también que además del citocromo c y en forma paralela, el Smac/DIABLO se libera de la mitocondria de las células HeLa infectadas, dando evidencias de la importancia de la vía mitocondrial de la apoptosis y específicamente la asociación del Smac/DIABLO con la inducción de apoptosis por el VLA. La acción sinérgica del citocromo c y el Smac/DIABLO son indicadores de la alteración en la permeabilidad de la membrana mitocondrial y llevan a la activación completa de las caspasas y a la muerte celular (17, 31). Consistentemente con nuestros datos, en la infección por reovirus se ha demostrado que la activación del Smac/DIABLO representa un evento mitocondrial clave en la inducción de apoptosis por reovirus en células mamíferas (19).
La liberación del citocromo c de la mitocondria juega un papel crucial en la cascada de la apoptosis a través de la activación de la procaspasa 9, por lo que es muy importante su detección para establecer la activación de la vía intrínseca de la apoptosis (22). La activación de la procaspasa 9 observada en células HeLa infectadas por el VLA, conjuntamente con la liberación de citocromo c es un hecho significante a tener en cuenta, ya que esta cascada de activación apoptótica se activaría exclusivamente en la ejecución del programa de apoptosis (28). Sin embargo, trabajos publicados (20, 32) indican que la pérdida del potencial de membrana de la mitocondria y la degradación del ADN puede también ocurrir bajo ciertas condiciones en células necróticas, por lo que no serían un indicador seguro de apoptosis. Por este motivo, para determinar apoptosis debemos analizar la activación de señales celulares, como el citocromo c y el Smac/DIABLO.
El NF-kB es un factor regulador tanto de genes pro- como anti-apoptóticos (2). En el presente trabajo hemos establecido una importante característica del proceso apoptótico en la infección por el VLA, que es la activación del NF-kB durante el primer ciclo viral en células HeLa. La activación de este factor fue detectada a las 12 y 18 h p.i. y la inhibición de la translocación nuclear del NF-kB por el uso del péptido SN50 indicó que este péptido bloquea el mecanismo intracelular de reconocimiento; la inhibición de la actividad del NF-kB se correlacionó con un máximo de 75% de reducción en la apoptosis celular. Este dato indica claramente que en la infección por el VLA se activa el NF-kB y que la inducción temprana de apoptosis es un efecto viral. Así, aunque en muchos casos la célula tendría como objetivo ejecutar el programa de apoptosis para prevenir la diseminación de la infección (30), en el caso del VLA el virus parecería tomar una ventaja de la inducción temprana de apoptosis en la activación de las caspasas. Esto se relaciona estrechamente con nuestras observaciones previas donde la actividad de las caspasas, en particular la caspasa 3, es requerida para llevar a cabo el proceso apoptótico. Así, podríamos puntualizar un fenómeno donde, en la infección por el VLA, el NF-kB actuaría como un factor pro apoptótico. Estos hallazgos se correlacionan con otras publicaciones donde se ha demostrado que en reovirus y flavivirus la inducción de apoptosis requiere la activación del NF-kB (10, 16, 23).
En el presente trabajo brindamos un aporte significativo para dilucidar el proceso biológico que ocurre en la célula mamífera luego de la infección por el VLA. Las experiencias llevadas a cabo suministran evidencia clara de la activación de las vías intrínseca y extrínseca de apoptosis en la infección por el VLA-10. Nuestros datos confirman el importante y en cierto modo crucial papel del NF-kB en la inducción de la apoptosis. El esclarecimiento de las vías de activación del proceso de apoptosis en la infección por el VLA es fundamental para conocer la forma en que el VLA causa la muerte celular. Continúa siendo un desafío a futuro definir precisamente el papel central de la apoptosis en la patogenia y diseminación del virus, así como también identificar otros mecanismos que se activan en la infección celular por el VLA.
Agradecimientos: debo expresar mi agradecimiento al Departamento de Enfermedades Tropicales Infecciosas de la Escuela de Higiene y Medicina Tropical de Londres, Inglaterra, donde se realizó parte de este trabajo durante mi estadía posdoctoral en la misma. Eduardo Mortola.
1. Akao M, ORourke B, Teshima Y, Seharaseyon J, Marban E. Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ Res 2003; 92: 186-94. [ Links ]
2. Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 1999; 18: 6910-24. [ Links ]
3. Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 1998; 17: 37-49. [ Links ]
4. Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science 1995; 267: 1485-8. [ Links ]
5. Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 2000; 406: 855-62. [ Links ]
6. Chen Z, Hagler J, Palombella V J, Melandri F, Scherer D, Ballard D et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev 1995; 9: 1586-97. [ Links ]
7. Clarke P, Tyler KL. Reovirus-induced apoptosis: A minireview. Apoptosis 2003; 8: 141-50. [ Links ]
8. Clarke P, Meintzer SM, Spalding AC, Johnson GL, Tyler KL. Caspase 8-dependent sensitization of cancer cells to TRAIL-induced apoptosis following reovirus-infection. Oncogene 2001; 20: 6910-9. [ Links ]
9. Clarke P, Meintzer, SM, Gibson, S, Widmann, C, Garrington, TP, Johnson GL, et al. Reovirus-induced apoptosis is mediated by TRAIL. J Virol 2000; 74: 8135-9. [ Links ]
10. Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, et al. Reovirus-induced apoptosis requires activation of transcription factor NF-kB. J Virol 2000; 74: 2981-9. [ Links ]
11. DeMaula CD, Leutenegger CM, Jutila MA, MacLachlan NJ. Bluetongue virus-induced activation of primary bovine lung microvascular endothelial cells. Vet Immunol Immunopathol 2002; 86: 147-57. [ Links ]
12. DeMaula CD, Jutila MA, Wilson DW, MacLachlan NJ. Infection kinetics, prostacyclin release and cytokine-mediated modulation of the mechanism of cell death during Bluetongue virus infection of cultured ovine and bovine pulmonary artery and lung microvascular endothelial cells. J Gen Virol 2001; 82: 787-94. [ Links ]
13. Duncan R, Muller J, Lee N, Esmaili A, Nakhasi HL. Rubella virus-induced apoptosis varies among cell lines and is modulated by Bcl-XL and caspase inhibitors. Virology 1999; 255: 117-28. [ Links ]
14. Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281:1309-12. [ Links ]
15. Jan JT, Griffin DE. Induction of apoptosis by Sindbis virus occurs at cell entry and does not require virus replication. J Virol 1999; 73: 10296-302. [ Links ]
16. Jan JT, Chen BH, Ma SH, Liu CI, Tsai HP, Wu HC, et al. Potential dengue virus-triggered apoptotic pathway in human neuroblastoma cells: arachidonic acid, superoxide anion, and NF-kB are sequentially involved. J Virol 2000, 74: 8680-91. [ Links ]
17. Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem 2004; 73: 87-106. [ Links ]
18. Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires both death receptor- and mitochondrial-mediated caspase-dependent pathways of cell death. Cell Death Differ 2002; 9: 926-33. [ Links ]
19. Kominsky DJ, Bickel RJ, Tyler KL. Reovirus-induced apoptosis requires mitochondrial release of Smac/DIABLO and involves reduction of cellular inhibitor of apoptosis protein levels. J Virol 2002; 76: 11414-24. [ Links ]
20. Lecoeur H. Nuclear apoptosis detection by flow cytometry: influence of endogenous endonucleases. Exp Cell Res 2002; 277: 1-14. [ Links ]
21. Lewis J, Oyler GA, Ueno K, Fannjiang YR, Chau BN, Vornov J, et al. Inhibition of virus-induced neuronal apoptosis by Bax. Nat Med 1999; 5: 832-5. [ Links ]
22. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479-89. [ Links ]
23. Liao CL, Lin YL, Wu BC, Tsao CH, Wang MC, Liu CI, et al. Salicylates inhibits flavivirus replication independently of blocking nuclear factor kappa B activation. J Virol 2001; 75: 7828-39. [ Links ]
24. Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003; 8: 115-28. [ Links ]
25. Mastrangelo AJ, Hardwick JM, Bex F, Betenbaugh MJ. Part I. Bcl-2 and Bcl-x (L) limit apoptosis upon infection with alphavirus vectors. Biotechnol Bioeng 2000; 67: 544-54. [ Links ]
26. Mortola E, Noad R, Roy P. Bluetongue virus outer capsid proteins are sufficient to trigger apoptosis in mammalian cells. J Virol 2004; 78: 2875-83. [ Links ]
27. Nagaleekar VK, Tiwari AK, Kataria RS, Bais MV, Ravindra PV, Kumar S. Bluetongue virus induces apoptosis in cultured mammalian cells by both caspase-dependent extrinsic and intrinsic apoptotic pathways. Arch Virol 2007; 152: 1751-6. [ Links ]
28. Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci 1997; 22: 299-306. [ Links ]
29. Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A, et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem 2003; 278: 28220-8. [ Links ]
30. Roulston A, Marcellus RC, Branton PE. Viruses and apoptosis. Annu Rev Microbiol 1999; 53: 577-628. [ Links ]
31. Salvesen GS, Duckett CS. IAP proteins: blocking the road to deaths door. Nat Rev Mol Cell Biol 2002; 3: 401-10. [ Links ]
32. Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 2000; 45: 528-37. [ Links ]
33. Sterin-Speziale N, Leocata Nieto F. Los esfingolípidos en la muerte y proliferación celular. Revista Química Viva 2007; 3: 234-50. [ Links ]
34. Van Loo G, Saelens X, Van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. The role of mitochondrial factors in apoptosis: a Russian roulette with more than one bullet. Cell Death Differ 2002; 9: 1031-42. [ Links ]
35. Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000; 102: 43-53. [ Links ]
36. Wang X. The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15: 2922-33. [ Links ]
37. Waterhouse NJ, Ricci JE, Green DR. And all of a sudden its over: mitochondrial outer-membrane permeabilization in apoptosis. Biochimie 2002; 84: 113-21. [ Links ]
38. Liu Y, Yinghui P, Xuming Z. Role of the mitochondrial signaling pathway in murine coronavirus-induced oligodendrocyte apoptosis. J Virol 2006; 80: 395-403. [ Links ]
39. Yin XM. Bid, a critical mediator for apoptosis induced by the activation of Fas/TNF-R1 death receptors in hepatocytes. J Mol Med 2000; 78: 203-11. [ Links ]
Recibido: 13/01/09
Aceptado: 14/07/09

