










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkRevista argentina de microbiología
versión impresa ISSN 0325-7541
Rev. argent. microbiol. vol.42 no.4 Ciudad Autónoma de Buenos Aires oct./dic. 2010
MICROBIOLOGÍA INDUSTRIAL Y AMBIENTAL
Estudio comparativo de la estructura del bacterioplancton en aguas del Mar Argentino mediante el método de pirosecuenciación 454 tag
S. R. Peressutti1 *, M. Costagliola1, L. F. Artigas2, C. Hozbor1
1 Instituto Nacional de Investigación y Desarrollo Pesquero (INIDEP) Paseo Victoria n° 1 (7600) Mar del Plata, Argentina,
2 Université Lille Nord de France, Université du Littoral Côte d'Opale, CNRS UMR 8187 LOG, Maison de la Recherche en Environnement Naturel, 32 av. Foch, 62930 Wimereux, France.
*Correspondencia. E-mail: silviap_ar@yahoo.com
RESUMEN
El presente estudio brinda la primera información sobre diversidad y abundancia de las comunidades microbianas en dos ambientes del Mar Argentino obtenida mediante la técnica de pirosecuenciación tag ribosomal 454. Dentro del dominio Bacteria, se observaron más de 4 600 secuencias únicas a partir de 36 188 amplicones de tags y se identificaron 280 filotipos. Además, se detectaron cerca de 2 700 secuencias únicas a partir de más de 47 700 tags pertenecientes al dominio Archaea, lo que definió sólo 5 filotipos diferentes. La distancia de Jaccard presentó valores de 0,6 para bacterias y de 0,2 para arqueas, esto indica mayor diferencia entre las bacterias en los dos sitios. En el ambiente marino los filotipos más dominantes fueron Bacteroidetes Flavobacteriaceae, Proteobacteria Gammaproteobacteria, Proteobacteria Rhodobacteraceae y Proteobacteria Rickettsiales SAR11, mientras que en el estuario predominaron Pseudoalteromonadaceae Pseudoalteromonas, Proteobacteria Gammaproteobacteria, Proteobacteria Shewanella y Proteobacteria Rickettsiales SAR11. Los 2 filotipos de arqueas encontrados en mayor proporción fueron Archaea Euryarchaeota y Archaea Crenarchaeota. Las secuencias tag más numerosas representaron taxa caracterizados previamente, aunque también se halló un elevado número de filotipos de gran diversidad y de baja abundancia, que forman parte de la denominada "biosfera rara", aún no explorada, que pueden tener un papel ecológico crucial.
Palabras claves: Diversidad microbiana; Mar Argentino; Pirosecuenciación tag ribosomal 454
ABSTRACT
A comparative study of bacterioplankton structure in Argentinian Sea waters by the 454 - tag pyrosequencing method. The present study provides the first information about diversity and abundance of microbial communities in two environments of the Argentinian Sea by the 454 - tag pyrosequencing technique. We observed more than 4,600 unique bacterial sequences from 36,188 tag amplicons, forming 280 phylotypes. In addition, nearly 2,700 unique sequences from more than 47,700 tags identified as Archaea, defined only 5 different phylotypes. The Jaccard distance (0.6 for Bacteria and 0.2 for Archaea) indicated higher differences among Bacteria rather than among Archaea in both studied sites. The dominant phylotypes in marine environment were Bacteroidetes Flavobacteriaceae, Proteobacteria Gammaproteobacteria, Proteobacteria Rhodobacteraceae and Proteobacteria Rickettsiales SAR11; and Pseudoalteromonadaceae Pseudoalteromonas, Proteobacteria Gammaproteobacteria, Proteobacteria Shewanella, Proteobacteria Rickettsiales SAR11 in the estuary sampling site. Archaea Euryarchaeota and Archaea Crenarchaeota were the major archaeal phylotypes found. The most abundant tag sequences included previously characterized taxa, although we also retrieved a large number of highly diverse, low-abundant phylotypes which constitute a largely unexplored "rare" biosphere. These microorganisms could have a crucial ecological role.
Key words: Microbial diversity; Argentinian Sea; 454 - tag ribosomal pyrosequencing
INTRODUCCIÓN
El bacterioplancton tiene un papel significativo como principal contribuyente en los procesos biogeoquímicos de los ambientes marinos y estuariales, y representa un componente fundamental en las cadenas alimentarias microbianas (36). En los últimos años, el concepto de la cadena trófica clásica ha cambiado a un modelo alternativo más complejo denominado microbial loop o "bucle microbiano" (4), basado en la capacidad de las bacterias heterótrofas de reciclar la materia orgánica disuelta y de hacerla circular mediante diversas interacciones ecológicas a través de los otros componentes del plancton y, en forma indirecta, también a través de los niveles tróficos superiores del ecosistema marino. En los océanos, los microorganismos son los responsables del 98% de la producción primaria e intervienen en todos los ciclos de la materia (40), con un papel fundamental, a su vez, en el destino de los contaminantes en zonas altamente antropizadas. Siguiendo el esquema trófico tradicional, el plancton es utilizado como alimento por los organismos de niveles tróficos más elevados (9). En consecuencia, la dinámica de los componentes del plancton podría explicar algunas pautas de comportamiento y distribución de organismos de niveles tróficos superiores, como por ejemplo, las variaciones espacio-temporales observadas en ciertas poblaciones de peces y sus larvas (23, 25).
El conocimiento de la estructura de la comunidad microbiana, que incluye la diversidad y la abundancia total y relativa, y de su relación con los factores ambientales es de gran importancia para comprender el papel de las bacterias en los ciclos biogeoquímicos y detectar posibles patrones predecibles, ya que esto permitiría un manejo más efectivo de los ecosistemas marinos (11).
Los estudios moleculares que comparan secuencias génicas de microorganismos han demostrado una gran diversidad bacteriana, al menos 100 veces mayor que las estimaciones basadas en técnicas dependientes del cultivo (26). Dentro de las técnicas moleculares utilizadas, las de fingerprinting como T-RFLP (Terminal-Restriction Fragment Length Polymorphism) y DGGE (Denaturing Gradient Gel Electrophoresis) permiten analizar la dinámica de la estructura de las comunidades en muestras ambientales (15).
En los últimos años se han incorporado técnicas a gran escala como las metagenómicas, basadas en el análisis de fragmentos genómicos de microorganismos del ambiente (41), y dentro de ellas, el uso de metodologías alternativas de secuenciación, como la pirosecuenciación (tecnología 454, Roche), para el estudio más detallado de la diversidad de comunidades microbianas naturales (20). Mediante estas técnicas se obtienen secuencias génicas que proveen una base para la estimación de la diversidad filogenética y generan "inventarios taxonómicos" de las poblaciones microbianas marinas (12). En trabajos anteriores se realizaron estudios de la riqueza del bacterioplancton a través de comparaciones del ARNr, y se encontraron desde unos pocos cientos de filotipos por ml en la columna de agua (38, 41) hasta 3 000 en sedimentos marinos (18).
El método de pirosecuenciación ribosomal basado en la secuenciación y el análisis de cientos de tags (pequeños fragmentos de alrededor de 100 pb de los genes 16S ARNr) brinda una información muy detallada de la diversidad de las comunidades, ya que permite detectar y cuantificar tanto los miembros más abundantes de la comunidad como aquellos componentes minoritarios, que son escasamente recuperados por cultivos tradicionales o clonación y secuenciación (6). Esta gran diversidad de microorganismos raros, también llamada "biosfera rara" (40), puede tener un papel importante en el ambiente acuático al constituir un "banco de reserva", con el potencial de volverse dominantes en respuesta a cambios en las condiciones ambientales, como por ejemplo, en la temperatura y la salinidad (28).
Debido a la escasa información existente acerca de la composición de las comunidades microbianas en ambientes acuáticos del área latinoamericana del Océano Atlántico, el proyecto International Census of Marine Microbes (ICoMM) ha concentrado sus esfuerzos en incrementar la eficiencia de los estudios de taxonomía microbiana basados en pirosecuenciación, que se aplican al estudio de aguas oceánicas y bentos en todo el planeta. En el marco de este proyecto se llevó a cabo un estudio comparativo de la composición y abundancia relativa de comunidades microbianas mediante el método de pirosecuenciación tag ribosomal 454 en dos ambientes costeros argentinos: una estación de muestreo del estuario del Río de la Plata y una estación marina permanente de estudios ambientales. El estuario del Río de la Plata se caracteriza por su gran extensión espacial (3,2 millones de km2), por su estratificación salina y por la presencia de una cuña o frente salino de fondo. Como una característica distintiva de este estuario y debido a las condiciones particulares de circulación de las aguas, la cuña salina genera un sistema frontal donde se producen diversos procesos ecológicos, por la acumulación de altas concentraciones de detrito y biomasa planctónica (37). En cambio, el sitio marino estudiado es un ambiente oligotrófico y poco estratificado (39). Esta estrategia de identificación fue utilizada con la finalidad de caracterizar la diversidad de bacterias y arqueas en dos zonas de la plataforma del Mar Argentino. Además, se analizó la abundancia total de bacterias y los parámetros ambientales que caracterizan los sitios estudiados.
MATERIALES Y MÉTODOS
Muestreos
Las muestras de agua fueron colectadas en el invierno del año 2006, en 2 muestreos realizados durante 2 campañas de investigación del INIDEP. Una de las muestras fue tomada en una estación permanente de estudios ambientales (EPEA, 38°28´S 57°41´W) y la otra en una estación de muestreo en la zona del estuario del Río de la Plata (n.° 444, 36°10,9´S 55°23,9´W) (Figura 1). Se colectaron muestras superficiales de agua en botellas estériles acondicionadas a 4 °C en oscuridad.
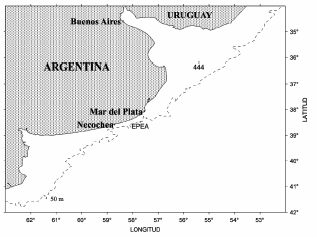
Figura 1. Ubicación de las posiciones de muestreo
Análisis físico-químicos
Las mediciones de temperatura y conductividad se realizaron en la columna de agua con un CTD SBE (Sea-Bird Electronic) modelo XIX, con fluorómetro (Seapoint Chlorophyll Fluorometer) adosado para la determinación in situ de los perfiles verticales de fluorescencia del fitoplancton in vivo. Para la determinación de la concentración de clorofila a se filtraron alícuotas de las muestras de agua a través de filtros de fibra de vidrio (Whatman GF/F), se almacenaron a -80 °C y luego se realizaron las mediciones mediante la técnica de espectrofluorometría (22).
Recuentos de bacterias totales
Se tomaron 100 ml de agua y se colocaron en colectores estériles, fijados con formaldehído (2%, vol/vol) y mantenidos en oscuridad a 4 °C. A partir de estas muestras se tomaron alícuotas de 2 ml y se tiñeron con DAPI (4'6- diamino-2-fenil-indol, 5 μg/ml) y con OA (naranja de acridina, 1 μg/ml) (19). Se utilizaron los dos fluorocromos para poder diferenciar las células bacterianas del background. La abundancia de bacterias totales se determinó por epifluorescencia utilizando un microscopio invertido Olympus IX70. Los recuentos de las células se realizaron mediante un programa informático de análisis de imágenes (Image Pro versión 4.0).
Extracción de ADN y purificación
En los análisis de diversidad se utilizaron las mismas muestras para la identificación de bacterias y de arqueas. Se prefiltraron 1,5 l de las muestras de agua a través de filtros GF/F de fibra de vidrio (Whatman) y luego por filtros Durapore (Millipore) de 0,22 μm, los cuales fueron enjuagados con buffer fosfato (34) y conservados en termos de nitrógeno líquido.
La extracción de ADN a partir de los filtros Durapore fue realizada utilizando fenol-cloroformo-alcohol isoamílico, tras haber efectuado una digestión enzimática mediante lisozima y proteinasa K (7). El ADN genómico fue cuantificado utilizando el colorante Hoechst 33258 (Amersham Biosciences, Piscataway, NJ) en un fluorómetro Hoefer DyNA quant 200 (Hoefer Scientific Instruments, San Francisco, CA).
Construcción de genotecas de amplicones de PCR y secuenciación tag 454
Las muestras de ADN fueron enviadas al Marine Biological Laboratory de Woods Hole, Massachusetts, EE.UU. Durante este estudio se utilizó la estrategia de secuenciación tag combinando el uso de amplicones de la región hipervariable V6 del ARNr 16S de la subunidad ribosomal menor para la identificación de filotipos individuales u OTU (operative taxonomic units, unidades taxonómicas operativas) mediante secuenciaciones masivas paralelas (16). A su vez, las secuencias conservadas que flanquean la región V6 de ARNr sirven como sitios de unión de los cebadores para generar los amplicones por PCR. Esta estrategia de secuenciación brinda la colección concurrente de 10 000-50 000 tags a partir de las muestras en una sola corrida de secuenciación de 4 horas (http://vamps.mbl.edu/).
Para la secuenciación de la región V6 de bacterias se usó una mezcla de 5 cebadores forward (967F): CNACGCGAAGAACCTTANC, CAACGCGAAAAACCTTACC, CAACGCGCAGAACC TTACC, ATACGCGARGAACCTTACC y CTAACCGANGAACCTYACC y 4 cebadores reverse (1046R): CGACAGCCATGCANCACCT, CGACAACCATGCANCACCT, CGACGGCCATGCANCACCT y CGACGACCATGCANCACCT. Para la secuenciación de la región V6 de arqueas se usó un cebador forward (958F): AATTGGANTCAACGCCGG y 2 cebadores reverse (1048R): 1048arcR-mayor CGRCGGCCATGCACCWC y 1048arcR-menor CGRCRGCCATGYACCWC. De este modo se generaron genotecas de amplicones de PCR para cada muestra de ADN ambiental. La mezcla de amplificación incluyó 5 unidades de Pfu turbo ADN polimerasa (Stratagene, La Jolla, CA), 1X de buffer de reacción, 200 μM de dNTP (Pierce Nucleic Acid Technologies, Milwaukee, WI) y 0,2 μM de cada cebador en un volumen de 100 μl. Se adicionaron 10 ng de ADN genómico (templado) a las mezclas de amplificación. Las condiciones de ciclado fueron las siguientes: una desnaturalización inicial a 94 °C durante 2 min; 30 ciclos de 94 °C 30 s, 50 °C 20 s y 72 °C 1 min; y una extensión final de 2 min a 72 °C. Se realizó un pool de los productos y éste fue purificado mediante el equipo de purificación MiniElute (Qiagen, Valencia, CA) y eluido en 30 μl de buffer EB. La calidad, el tamaño y la concentración del producto de PCR fueron ensayados sobre un Bioanalyzer 2100 (Agilen, Palo Alto, CA) usando un DNA1000 LabChip. Sólo los productos de amplificación diferentes y con una producción total > 200 ng fueron utilizados para la secuenciación 454. Los fragmentos en las genotecas de amplicones fueron unidos a esferas o beads, y éstas fueron emulsionadas en una mezcla de PCR en aceite; así se generaron alrededor de 10 millones de copias de un único templado de ADN. Luego de la ruptura de la emulsión, las hebras de ADN fueron desnaturalizadas, y las esferas que llevaban clones de ADN de una hebra fueron depositadas en calles sobre un PicoTiter-Plate (454 Life-Sciences) para pirosecuenciación, sobre un sistema Genome Sequencer FLX (Roche, Basilea, Suiza) en 454 Life Sciences (Branford, CT). Las secuencias obtenidas fueron sometidas a un proceso de selección con el fin de minimizar los efectos de errores de secuenciación al azar. Durante ese proceso se eliminaron las secuencias tag de baja calidad que presentaron una o más bases ambiguas, las que tuvieron un largo menor de 50 nucleótidos y las que no coincidieron con los cebadores de la PCR al inicio de la lectura.
Asignación taxonómica de filotipos (OTU)
Para asignar una clasificación taxonómica a las secuencias tag-454 V6 ambientales se usó el proceso GAST (Global Alignment for Sequence Taxonomy), que constituye una metodología de mapeo de las tags (http://vamps.mbl.edu/). Se creó una base de datos de referencia de genes ARNr, RefSSU, según la base de datos SILVA (32), y una base de datos de referencia (V6RefDB) de la región tag hipervariable V6 a partir de RefSSU usando también el alineamiento SILVA. Mediante BLAST (Basic Local Alignment Search Tool) se comparó cada tag de pirosecuenciación con la base de datos de referencia V6RefDB para generar un conjunto de 100 mejores uniones. Se utilizó el programa MUSCLELE (8) para alinear las secuencias tag a las tags de referencia correspondiente a las 100 mejores BLAST.
Se identificó el total de las secuencias de referencia en Ref16S que contenían la secuencia hipervariable exacta de las mejores asociaciones GAST. Si dos tercios o más de la longitud total de la secuencia compartía el mismo género asignado, la secuencia tag fue clasificada a nivel género; de lo contrario, se continuó a nivel de familia o a otro nivel taxonómico. Así, las asignaciones de filotipos se realizaron de acuerdo con las secuencias de la región V6 de referencia que presentaron la mínima distancia con la secuencia problema. La frecuencia de las mejores asociaciones a V6RefDB para cada sitio fue usada para calcular las curvas de rarefacción con el programa Analytic rarefaction 1.3. Los análisis de rarefacción permiten estimar si el número de secuencias obtenidas representan una fracción significativa de las muestras (2, 30). La cobertura de los respectivos pools de secuencias fue calculada usando los estimadores de Good (13) y Chao (21). Asimismo, se comparó la biodiversidad entre bacterias y entre arqueas en los dos ambientes estudiados utilizando la distancia de Jaccard como medida de disimilitud.
Los datos fueron incorporados en el sitio web VAMPS (http://vamps.mbl.edu/), que sirve como herramienta para medir y visualizar similitudes y diferencias entre los perfiles moleculares de comunidades microbianas complejas. Actualmente, VAMPS se centraliza en el análisis de conjuntos de datos masivos de secuencias cortas e hipervariables de genes ARNr (V6-ARNr tags) a partir de poblaciones microbianas naturales. En un futuro, se extenderá a comparaciones de comunidades basadas en el largo total de ARNr, y finalmente, a datos metagenómicos (secuencias colectivas de genomas en una comunidad microbiana mixta).
Finalmente, con el fin de evaluar las asignaciones taxonómicas de tags hipervariables vía GAST elegimos 100 secuencias al azar, que fueron exitosamente asignadas a nivel de orden por el VAMPS y las comparamos con la base de datos del RDP (Ribosomal Data Project) y con la del GenBank.
RESULTADOS
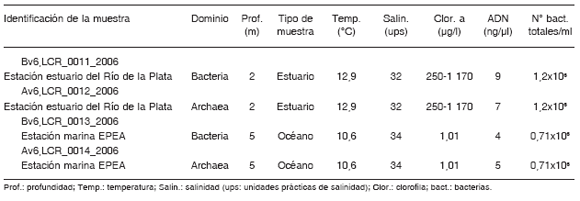
La estación EPEA y el sitio de muestreo del estuario mostraron condiciones de salinidad, temperatura y clorofila a típicas de las zonas marina y estuarial en la época invernal (Tabla 1). Se obtuvieron cantidades suficientes de ADN para ser amplificadas con cebadores específicos para bacterias y arqueas y, en ambos sitios, los recuentos bacterianos estuvieron en el orden de 106.
Tabla 1. Parámetros biológicos y físico-químicos de los sitios de muestreo.
Los análisis de biodiversidad fueron realizados mediante la secuenciación de productos de PCR de la región V6 (subunidad ribosomal menor) de arqueas y bacterias, a partir de las extracciones de ADN de las muestras de agua correspondientes a los dos sitios analizados. Luego de la secuenciación se realizó el proceso de selección donde se descartaron entre el 10% y el 15% de las lecturas iniciales, con lo cual se obtuvo un total de 83 902 secuencias (Tabla 2). A su vez, todas las secuencias iguales fueron agrupadas en 7 397 secuencias tag V6 únicas. Estas fueron comparadas con la base de datos de referencia (V6RefDB) y, en la mayoría de los casos, la información de las regiones tag V6 fue suficiente para lograr una caracterización filogenética. Los datos obtenidos fueron volcados en el sitio web VAMPS.
Por otro lado, se realizó una comparación de las asignaciones del VAMPS con la base de datos RDP (datos no mostrados), y se observó que muchas secuencias fueron clasificadas en forma distinta al VAMPS, tanto a nivel de phylum como de clase u orden. A nivel de phylum, las asignaciones diferentes representaron el 20%, mientras que a nivel de orden, el 54%. Cabe destacar que las secuencias asignadas por el RDP diferentes al VAMPS, en general, tuvieron porcentajes de confianza de un 20%, si bien entre las que se asignaban igual que el VAMPS había casos con valores bajos de confianza, por lo cual el RDP no resultaría un buen control en este análisis. En contraste, al analizar las secuencias que habían sido "mal asignadas" por el RDP utilizando BLAST, se determinó que en casi todos los casos la asignación coincidía con el VAMPS, hecho que demuestra la eficiencia de esta herramienta de identificación.
Tabla 2. Número de secuencias y filotipos de las muestras
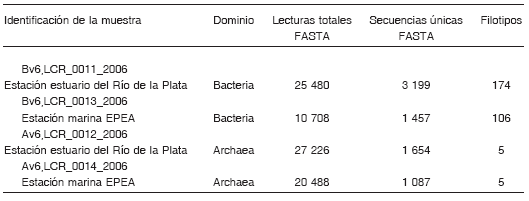
Como se observa en la Tabla 2, dentro del dominio Bacteria se obtuvieron más de 4 600 secuencias únicas a partir de 36 188 lecturas totales; esto generó 280 secuencias tag definidas como filotipos diferentes de acuerdo a sus coincidencias con las secuencias en la base de datos V6RefDB. En cuanto al dominio Archaea, se identificaron 2 741 secuencias únicas a partir de 47 714 secuencias tag, y finalmente se definieron sólo 5 filotipos diferentes. Además, se determinó un mayor número de secuencias de bacterias y arqueas en el estuario que en el ambiente marino. Las curvas de rarefacción se calcularon sobre la base de las mejores equivalencias entre cada tag y las secuencias en V6RefDB considerando 3 niveles de diferencias taxonómicas (3%, 6% y 10% a nivel de especie, de familia y de phylum, respectivamente; Fig. 2). Como se observa en la Figura 2 A y B, para el dominio Bacteria los valores no se vuelven constantes, sino que se observan curvas que no alcanzaron totalmente la fase plateau, mientras que en el caso de las arqueas las pendientes se vuelven casi asintóticas (Fig. 2 C y D), especialmente al considerar las secuencias que no excedieron el 3% y el 6% de diferencia con respecto a las bases de referencia. Se utilizaron los estimadores no paramétricos de Good y Chao para medir la cobertura de secuencias y la heterogeneidad de las poblaciones microbianas, combinando el número de diferentes tipos de organismos y sus abundancias relativas. Los resultados de estas estimaciones indicaron que el muestreo permitiría detectar el 97% de la riqueza de OTU en ambos ambientes.
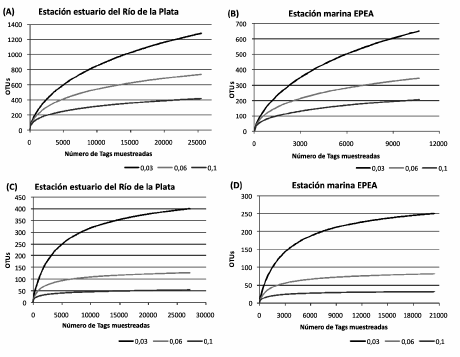
Figura 2. Curvas de rarefacción de bacterias (A y B) y arqueas (C y D) basadas en las mejores asociaciones con la base de datos V6RefDB, considerando diferencias del 3%, 6% y 10%.
Por otro lado, al analizar las secuencias se observó que la mayoría coincide con la base de datos de referencia (V6RefDB) y se encontró un alto número de tags iguales. De este modo, las 10 secuencias totales más abundantes se repitieron de 206 a 2 491 veces en bacterias y de 185 a 7 078 en arqueas, y concordaron exactamente con las secuencias de la base de datos (datos no mostrados). Además, de las 36 188 secuencias totales de bacterias halladas en los dos sitios (Tabla 2), el 77% estuvieron representadas por 100 secuencias únicas; en cambio, en arqueas, de las 47 714 secuencias totales, el 86% estuvieron representadas por 50 secuencias únicas, lo que muestra la mayor diversidad del dominio Bacteria. Como se observa en la Figura 3, el 76% y el 65% de secuencias totales y el 15% y el 3% de secuencias únicas son idénticas a las regiones V6RefDB para bacterias y arqueas, respectivamente. La mayor proporción de secuencias únicas de bacterias y arqueas se diferenciaron de 1% a 3% con las de referencia; alrededor del 15% de las secuencias únicas difirieron entre un 4% y un 10%, y en el caso de bacterias, cerca de un 1,5% se diferenciaron más del 10%.
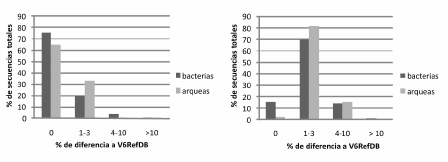
Figura 3. Similitud de las secuencias tag 454 totales (izquierda) y únicas (derecha) con la base de referencia V6RefDB.
Asimismo, un alto número de secuencias únicas de bacterias que difirieron entre 1% y 3% y, más aún, aquellas que se diferenciaron en más del 4% fueron divergentes entre sí, ya que estuvieron representadas por sólo una o dos secuencias totales.
Los análisis taxonómicos permitieron caracterizar a las comunidades microbianas principalmente a nivel de género, mientras que sólo fue posible identificar un pequeño porcentaje a nivel de especie (Figura 4). Además, se comparó la biodiversidad mediante la distancia de Jaccard en los dos ambientes estudiados. Los valores obtenidos fueron de alrededor de 0,6 a nivel de especie y de orden para bacterias y de 0,2 para arqueas, lo que muestra mayores diferencias entre las bacterias que entre las arqueas, lo cual es evidente al comparar la estructura de las comunidades (Figuras 5, 6 y 7). En las Figuras 5 y 6 se muestra la clasificación de las secuencias tag únicas. Estas fueron asignadas a diferentes niveles taxonómicos, desde el nivel de especie hasta incluso en algunos casos sólo se clasificaron a nivel de dominio Bacteria. Entre las bacterias del sitio marino, los filotipos representados en una proporción mayor o igual al 5% incluyeron Bacteroidetes Flavobacteriaceae (30,4%), Proteobacteria Gammaproteobacteria (17,6%), Proteobacteria Rhodobacteraceae (17,4%), Proteobacteria Rickettsiales SAR11 (5%) y Bacteroidetes Flavobacteriales (5%), mientras que en el estuario predominaron Pseudoalteromonadaceae Pseudoalteromonas (22,4%), Proteobacteria Gammaproteobacteria (11,4%), Proteobacteria Shewanellaceae Shewanella (9,8%), Proteobacteria Rickettsiales SAR11 (7,4%), Bacteroidetes Cytophagaceae Sporocytophaga (5,9%) y Proteobacteria Pseudomonadaceae Pseudomonas (5,4%). También se encontraron filotipos en una frecuencia menor del 0,05%, como por ejemplo Proteobacteria Caulobacteraceae y Acidobacteria en el estuario, Flavobacteriaceae Polaribacter en el sitio marino o Cyanobacteria en ambos ambientes. A través de esta identificación de taxa también se determinó mayor diversidad de filotipos bacterianos en el estuario que en el ambiente marino.
En cuanto a las arqueas (Figura 7), la mayoría fueron clasificadas a nivel de phylum y una pequeña proporción se identificó sólo a nivel de dominio. Los 2 filotipos encontrados en mayor proporción fueron Archaea Euryarchaeota (99,8% en el océano y 64,9% en el estuario) y Archaea Crenarchaeota (0,14% en el océano y 33,3% en el estuario).
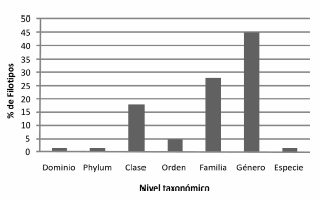
Figura 4. Porcentaje de OTU a diferentes niveles taxonómicos.
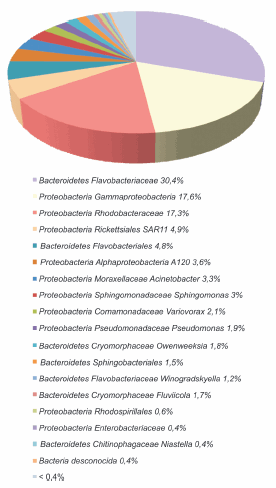
Figura 5. Diversidad de bacterias en la estación permanente EPEA. Las secuencias tag únicas fueron clasificadas a diferentes niveles taxonómicos.
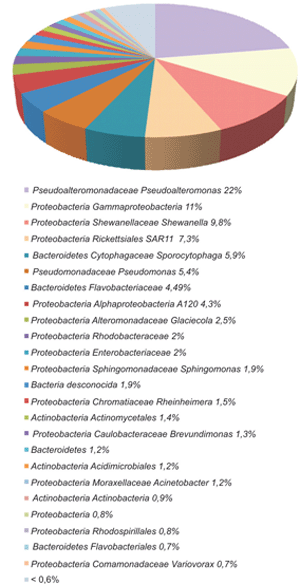
Figura 6. Diversidad de bacterias en la estación del estuario del Río de la Plata. Las secuencias tag únicas fueron clasificadas a diferentes niveles taxonómicos.
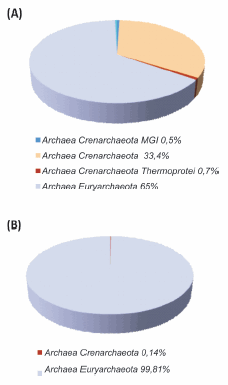
Figura 7. Diversidad de arqueas en la estación del estuario del Río de la Plata (A) y en la estación EPEA (B). Las secuencias tag únicas fueron clasificadas a diferentes niveles taxonómicos.
DISCUSIÓN
En este trabajo se analizó la diversidad microbiana y su abundancia relativa en dos ambientes dentro de la plataforma del Mar Argentino. Se detectaron condiciones ambientales típicas de los dos sitos analizados y los recuentos bacterianos, determinados mediante tinción con fluorocromos, fueron similares a los informados previamente en otras áreas marinas y estuariales (12, 31).
En general, existe escasa información acerca de la gran biodiversidad de bacterias en sistemas marinos (40), y particularmente en el Mar Argentino no hay antecedentes publicados sobre el tema. En el presente estudio, la diversidad microbiana fue analizada utilizando la capacidad de secuenciación masiva paralela de ADN que brinda la tecnología 454 Life Sciences (3). Esta estrategia de secuenciación tag permitió incrementar varios órdenes de magnitud el número de amplicones y a partir del análisis de las secuencias se pudo establecer una biodiversidad mayor que la reportada previamente en otros ambientes marinos (12, 15). Cabe destacar que es poco probable que la diversidad hallada pueda ser atribuida a errores de secuenciación al azar, ya que como se describió anteriormente (17), los exhaustivos controles de calidad minimizan los errores de la PCR y de pirosecuenciación (< 0,0025 por base) y de asignación de la secuencia a los filotipos (< 1%). Por otro lado, es importante tener en cuenta las limitaciones del método, ya que al igual que en estudios previos (16), la resolución taxonómica de la mayoría de las secuencias tag V6 para el dominio Bacteria permitió una discriminación a nivel de género, pero no de especie, mientras que en las arqueas sólo a nivel de phylum. Sin duda, este es el principal inconveniente de la presente metodología.
En este estudio se realizaron las curvas de rarefacción con la finalidad de exponer gráficamente la eficiencia del muestreo. En ellas, en general se observa que al principio del muestreo de tags una gran variedad de nuevos organismos son descubiertos, pero a medida que progresa el muestreo el número va disminuyendo hasta alcanzar una fase asintótica. Las curvas obtenidas en nuestro estudio no llegaron totalmente a un plateau, lo que da cuenta de la gran complejidad de la diversidad bacteriana existente en ambientes marinos (27). Sin embargo, el elevado número de secuencias únicas halladas indica una adecuada cobertura de muestreo para estas genotecas de secuencias. Al igual que en otros estudios (16), las curvas de rarefacción en arqueas se vuelven casi asintóticas, lo cual corrobora la eficiencia del muestreo para el estudio de la riqueza de arqueas en estos sistemas acuáticos.
El análisis de las secuencias recuperadas mostró un elevado número de tags idénticas y la mayoría concordaron exactamente con las de la base de datos de referencia o presentaron una divergencia nucleotídica menor del 1%, esto corrobora la representatividad del muestreo, especialmente para las arqueas. A su vez, se determinó un mayor número de secuencias totales y únicas de bacterias que de arqueas, y al comparar el número de secuencias totales con el de secuencias únicas se demostró que hubo mayor diversidad bacteriana, al igual que en otros reportes (16). Estos resultados fueron confirmados mediante el análisis de distancia de Jaccard. También se encontró un gran número de secuencias únicas de bacterias relacionadas con sólo una o dos secuencias totales, y muchas de estas secuencias divergentes se asociaron con filotipos "raros" no informados anteriormente.
Los análisis taxonómicos y estadísticos revelaron diferencias entre los filotipos de las comunidades bacterianas en los dos sitios estudiados, con mayor diversidad en el estuario que en el ambiente marino. Estos sistemas acuáticos poseen características distintivas; el estuario es un ecosistema biológicamente más productivo que el marino y particularmente el del Río de la Plata, que se caracteriza por la formación de una cuña salina, presenta áreas frontales donde se acumulan altas concentraciones de detrito, condición que favorece el aumento de diversidad y biomasa planctónica (37, 39).
El análisis taxonómico reveló que un gran número de las secuencias tag obtenidas en los dos sitios de muestreo representaron taxa de distribución ubicua, que ya han sido caracterizados previamente. La mayor parte de los filotipos predominantes estuvieron presentes en ambos ambientes, aunque cabe destacar que estos fueron encontrados en distinta proporción. Así, Bacteroidetes Flavobacteriaceae estuvo presente en un 30,4% en el sitio marino y en un 4,5% en el estuario; Proteobacteria Gammaproteobacteria en un 17,6% en aguas marinas y en un 11,4% en aguas estuariales; y Proteobacteria Rickettsiales SAR11 un 5% en la primeras y un 7,4% en las segundas. Proteobacteria Gammaproteobacteria y Proteobacteria Rickettsiales SAR11 han sido también hallados en diferentes sistemas acuáticos alrededor del mundo (30, 41). Otros taxa cosmopolitas como Proteo- bacteria Rhodobacteraceae y Pseudomonadaceae Pseudomonas se caracterizan por su versatilidad genómica y metabólica, que les permite sobrevivir en amplias condiciones ambientales (5, 29). Por otro lado, el filotipo encontrado en mayor proporción en el estuario fue Pseudoalteromonadaceae Pseudoalteromonas, a diferencia del sitio marino, donde sólo se recuperaron unas pocas secuencias pertenecientes a este taxón. Estos resultados se contraponen con estudios anteriores realizados por DGGE y por el análisis del 16s ARNr de cepas aisladas, en los que se describió este género en diversas áreas marinas, mientras que sólo se halló en forma conspicua en la zona estuarial del Mar Báltico (14, 33). Es posible que estas diferencias se relacionen con la metodología utilizada y con las características ambientales particulares de los sistemas estudiados. De todos modos, serían necesarios estudios adicionales sobre nuevas muestras de agua para demostrar fehacientemente la dominancia de dicho filotipo en este ambiente estuarial.
La fracción predominante de diversidad, conocida también como "microdiversidad" o microclusters, ha sido observada en otros estudios de comunidades microbianas marinas (1) y no se explica por errores de PCR o por copias ribosomales múltiples, y es independiente de la estrategia molecular utilizada (35). Es notable que estos grupos de "microdiversidad" podrían representar importantes unidades de diferenciación taxonómica dentro de una comunidad microbiana.
Además, la comparación de cada secuencia única con la base de datos de referencia de la región V6 reveló no sólo taxa bien conocidos, sino también -al igual que en otros trabajos (40) - filotipos microbianos "raros", algunos de ellos no descritos anteriormente. La diversidad de taxa de baja abundancia constituye una "biosfera rara" que no ha sido explorada (28), y sería de gran importancia su caracterización desde el punto de vista taxonómico y de distribución biogeográfica. Es posible que algunos de estos taxa posean una distribución ubicua, mientras que otros tal vez habiten ambientes específicos, como producto de selecciones funcionales.
En este trabajo se hallaron taxa cosmopolitas, pero también se identificaron filotipos endémicos propios de cada ambiente. Estos resultados se contraponen con la hipótesis propuesta por Fenchel y Finally (10), quienes postularon que los microorganismos planctónicos deberían ser cosmopolitas. Sin embargo, esta idea de "everything is everywhere" tampoco parece estar de acuerdo con otros estudios previos (30) en los que se demostró que los patrones de diversidad espacial están fuertemente asociados a factores ambientales.
En cuanto a las arqueas, el escaso número de filotipos diferentes hallados coincide con lo informado por otros autores, quienes consideran que la diversidad de arqueas es relativamente limitada en ambientes marinos y estuariales (24). Sin embargo, sería importante profundizar estos estudios ya que, como se describió anteriormente, mediante este método sólo pudieron ser clasificadas a nivel de phylum.
Finalmente, aunque en este estudio solo examinamos muestras de dos ambientes pelágicos de la costa del Mar Argentino, este trabajo ha sido importante para determinar la eficiencia del muestreo y la identificación exhaustiva de comunidades microbianas en ambientes acuáticos para los que no existe información previa. Además, un trabajo realizado en el Marine Biological Laboratory (Woods Hole, MA) y aún no publicado, en el que se analizaron varias muestras de un mismo sitio, reveló la reproducibilidad del método. Por otra parte, en los estudios clásicos moleculares sólo se analizan los taxa altamente abundantes. En cambio, la pirosecuenciación es una herramienta poderosa que nos permite evaluar en forma comparativa la diversidad microbiana y brinda información taxonómica no sólo de los filotipos que se encuentran en alta proporción, sino también de aquellos poco frecuentes, pero que pueden tener un papel ecológico determinante. Este método complementa otros estudios moleculares e implica un aporte fundamental para el mejor entendimiento del impacto de la diversidad microbiana sobre los ecosistemas marinos. El presente trabajo brinda una base para futuros estudios de biodiversidad, con la finalidad de comprender el papel de las bacterias en ecosistemas marinos y de detectar variaciones en la estructura de las comunidades en respuesta a cambios ambientales locales y globales.
Agradecimientos: agradecemos a la Dra. Linda Amaral-Zettler por su excelente trabajo en el análisis de secuenciación del ADN.
1. Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF. Fine-scale phylogenetic architecture of a complex bacterial community. Nature 2004; 430: 551-4. [ Links ]
2. Acinas SG. Diversidad y estructura de una comunidad microbiana. Actualidad SEM - Bol Inf Soc Esp Microb 2007; 44: 24-9. [ Links ]
3. Amaral-Zettler LA, McCliment EA, Ducklow HW, Huse SM. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes. PLoS ONE 2009; 4: e6372. [ Links ]
4. Azam F, Fenchel T, Field JG, Gray JS, Meyer-Reil LA, Thingstad F. The ecological role of water-column microbes in the sea. Mar Ecol Prog Ser 1983; 10: 257-3. [ Links ]
5. Bano N, Hollibaugh JT. Phylogenetic composition of bacterioplankton assemblages from the Arctic Ocean. Appl Environ Microbiol 2002; 68: 505-18. [ Links ]
6. Brown MV, Philip GK, Bunge JA, Smith MC, Bissett A, Lauro FM, et al. Microbial community structure in the North Pacific Ocean. Int Soc Microb Ecol 2009; 3: 1374-86. [ Links ]
7. Dumestre JF, Casamayor EO, Massana R, Pedrós-Alió C. Changes in bacterial and archaeal assemblages in an equatorial river induced by the water eutrophication of Petit Saut dam reservoir (French Guiana). Aquat Microb Ecol 2001; 26: 209-21. [ Links ]
8. Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004; 5: 113. [ Links ]
9. Fenchel T. Marine plankton food chains. Annu Rev Ecol System 1988; 19: 19-38. [ Links ]
10. Fenchel T, Finally BJ. The ubiquity of small species: patterns of local and global diversity. Bioscience 2004; 54: 777-84. [ Links ]
11. Fenchel T. The microbial loop-25 years later. J Exp Mar Biol Ecol 2008; 366: 99-103. [ Links ]
12. Giovannoni SJ, Sting U. Molecular diversity and ecology of microbial plankton. Nature 2005; 437: 343-8. [ Links ]
13. Good IL. The population frequencies of species and the estimation of population parameters. Biometrika 1953; 80: 193-201. [ Links ]
14. Hagström A, Pinhassi J, Zweifel UL. Biogeographical diversity among marine bacterioplankton. Aquat Microb Ecol 2000; 21: 231-44. [ Links ]
15. Hewson I, Fuhrman JA. Richness and diversity of bacterioplankton species along an estuarine gradient in Moreton Bay, Australia. Appl Environ Microbiol 2004; 70: 3425-33. [ Links ]
16. Huber JA, Welch DBM, Morrison HG, Huse SM, Neal PR, Butterfield DA, et al. Microbial population structures in the deep marine biosphere. Science 2007; 318: 97-100. [ Links ]
17. Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 2007; 8: R143. [ Links ]
18. Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microb Ecol 2004; 47: 161-77. [ Links ]
19. Kuwae T, Hosokama Y. Determination of abundance and biovolume of bacteria in sediments by dual staining with 4',6-diamidino-2-phenylindole and acridine orange: relationship to dispersion treatment and sediment characteristics. Appl Environ Microbiol 1999; 65: 3407-12. [ Links ]
20. Kysela DL, Palacios C, Sogin ML. Serial Analysis of V6 ribosomal sequence tags (SARST-V6): A method for efficient, high-throughput analysis of microbial community composition. Environ Microbiol 2005; 7: 356-64. [ Links ]
21. Lee SM, Chao A. Estimating population size via sample coverage for closed capture-recapture models. Biometrics 1994; 50: 88-97. [ Links ]
22. Lutz V, Carreto JI. A new spectroflurometric method for the determination of chlorophylls and degradation products and its application in two frontal areas of the Argentine Sea. Cont Shelf Res 1991; 11: 433-51. [ Links ]
23. Marrari M, Viñas MD, Martos P, Hernández D. Spatial patterns of mesozooplancton distribution in the Southwestern Atlantic Ocean (34°-41°S) during austral spring: relationship with the hydrographic conditions. ICES J Mar Scien 2004; 61: 667-79. [ Links ]
24. Massana R, Murray AE, Preston CM, DeLong EF. Vertical distribution and phylogenetic characterization of marine planktonic Archaea in the Santa Barbara Channel. Appl Environ Microbiol 1997; 63: 50-6. [ Links ]
25. Munk P, Hansen BW, Nielsen TG, Thomsen HA. Changes in plankton and fish larvae communities across hydrographic fronts off West Greenland. J Plankton Res 2003; 25: 815-30. [ Links ]
26. Pace NR. A molecular view of microbial diversity and the biosphere. Science 1997; 274: 734-40. [ Links ]
27. Pedrós-Alió C. Diversity of bacterioplankton. Trends Ecol Evol 1993; 8: 86-90. [ Links ]
28. Pedrós-Alió C. Marine microbial diversity: can it be determined? Trends Microbiol 2006; 14: 257-63. [ Links ]
29. Pinhassi J, Montserrat Sala M, Havskum H, Peters F, Guadayol O, Malits A, et al. Changes in bacterioplankton composition under different phytoplankton regimens. Appl Environ Microbiol 2004; 70: 6753-66. [ Links ]
30. Pommier T, Canbäck B, Riemann L, Boström KH, Simu K, Lundberg P, et al. Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 2007; 16: 867-80. [ Links ]
31. Porter KG, Feig YS. The use of DAPI for identifying and counting aquatic microflora. Limnol Oceanogr 1980; 25: 943-8. [ Links ]
32. Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl Acids Res 2007; 35: 7188-96. [ Links ]
33. Riemann L, Leitet C, Pommier T, Simu K, Holmfeldt K, Larsson U, et al. The native bacterioplankton community in the Central Baltic Sea is influenced by freshwater bacterial species. Appl Envir Microbiol, 2008; 74: 503-15. [ Links ]
34. Rivera I, Lipp E, Gil A., Choopun N, Huq A, Colwell R. Method for extraction and application of Multiplex PCR to detect toxigenic V. cholerae O1 and O139 from aquatic ecosystems. Environ Microbiol 2003; 5: 599-606. [ Links ]
35. Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 2007; 5: e77. [ Links ]
36. Schauer M, Balagué V, Pedros-Alio C, Massana R. Seasonal changes in the taxonomic composition of bacterioplankton in a coastal oligotrophic system. Aquat Microb Ecol 2003; 31: 163-74. [ Links ]
37. Schiariti A, Berasategui A, Gilberto D, Guerrero R, Acha M, Mianzan H. Living in the front: Neomysis americana (Mysidacea) in the Río de la Plata estuary, Argentina-Uruguay. Mar Biol 2006; 149: 483-9. [ Links ]
38. Schloss PD, Handelsman J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 2005; 71: 1501-6. [ Links ]
39. Silva R, Negri R, Lutz V. Summer succession of ultraphytoplankton at the EPEA coastal station (Northern Argentina). J Planckton Res 2009; 32: 1-12. [ Links ]
40. Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta et al. Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc Natl Acad Sci USA. 2006; 103: 12115-20. [ Links ]
41. Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004; 304: 66-74. [ Links ]
Recibido: 04/05/10
Aceptado: 16/08/10

