











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkDesarrollo de la secuenciación masiva
En 1975, Sanger y Coulson43publicaron el primer método enzimático para secuenciar el ADN a través de la incorporación de dideoxinucleótidos terminales, y poco después secuenciaron por primera vez un genoma, el del bacteriófago MS2 o Phi-X17442. Una década más tarde aparecieron los secuenciadores automáticos, que primero empleaban geles (Applied Biosystems PRISM® 373) y después capilares recubiertos de polímero (ABI PRISM® 310), y en 1995 se completó la secuencia del primer genoma bacteriano, el de Haemophilus influenzae18. En la actualidad existen casi 120.000 genomas de especies bacterianas distintas depositados en diversas bases de datos, como PATRIC (www.patricbrc.org/view/DataType/Genomes, consultado a 10 de diciembre de 2018).
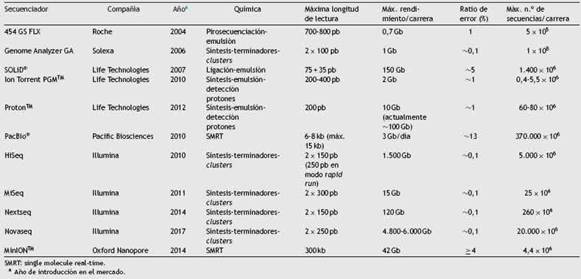
A comienzos del siglo XXI surgen nuevos métodos de secuenciación, basados en la pirosecuenciación y las denominadas plataformas de Next Generation Sequencing (NGS) que popularizó la compañía Life Sciences-Roche con su equipo 454 GS, comercializado por primera vez en 200436. Hoy en día, es más apropiado hablar de High-Throughput Sequencing (HTS) o secuenciación masiva, porque han surgido nuevas generaciones de secuenciadores que aplican otras tecnologías de secuenciación en paralelo. Entre estas se encuentra la secuenciación por ligación (Sequencing by Oligonucleotide Ligation and Detection) del equipo SOLiD ―introducido en el mercado en 2007 por Life Technologies y ya descatalogado―, la secuenciación por síntesis y semiconducción del Ion Torrent ―tecnología que fue adquirida por Life Technologies―, la secuenciación por síntesis en clusters de la empresa Solexa ―luego, Illumina―, que apareció en su primer equipo GAII, de 2006, y que es utilizada en los demás secuenciadores que fueron comercializados posteriormente, como el MiSeq, a partir de 2011 (este es el secuenciador más adecuado para microbiología, puesto que es el único secuenciador de mesa de laboratorio con una longitud de hasta 600pb, con paired end sequencing y exactitud de lectura de más del 99,9%). Otros secuenciadores basados en esa tecnología son HiSeq, NextSeq, NovaSeq y MiniSeq. Una tercera generación de secuenciadores son los que usan la secuenciación de molécula única (single molecule real-time [SMRT]), como el equipo portátil MinION( de Oxford Nanopore Technologies (2014) o el equipo PacBio® de Pacific Biosciences (2010), que permiten secuenciar moléculas mucho más largas, de hasta 30kb. La tabla 1 tabla 1 recoge las características de los principales secuenciadores masivos34,47.
La aplicación de la secuenciación masiva, además de reducir los costes y el tiempo de análisis, genera una gran cantidad de información, que está cambiando el modo de cómo se realiza la investigación en microbiología, y demanda, inexorablemente, la aplicación de la bioinformática en el diagnóstico y el análisis microbiológico. De hecho, en genómica no se cumple la ley de Moore, ya que se duplica la capacidad de secuenciar cada 6 a 9 meses (y no cada 2 años, como ocurre con los microprocesadores). La bioinformática aplica las matemáticas, la estadística y la computación al estudio biológico, por tanto, requiere de unos conocimientos básicos en lenguajes de programación (Bash, Perl, Python y R, entre otros) y, preferentemente, también en el uso de sistemas operativos basados en UNIX®, como Linux y MacOS®. Al ser de licencia y código libres, Linux se ha convertido en la opción de preferencia para los investigadores. Las distintas distribuciones de Linux (Ubuntu, CentOS, Mint, etc.) ofrecen interfaces sencillas para el usuario, que recuerdan a aquellas de los ordenadores de oficina. Sin embargo, el verdadero poder de estas plataformas reside en el uso de la terminal, que requiere, a su vez, conocimiento del lenguaje Bash como intérprete de comandos, de tal forma que se concentre todo el potencial del computador en la tarea que se quiera realizar.
La variedad y cantidad de programas informáticos que proporcionan una interfaz gráfica son limitados y le restan poder computacional al desarrollarla, por lo que se vuelve necesario el uso de programas ejecutados desde la terminal. Aunque se trata de una disciplina nueva para muchos microbiólogos, la comunidad científica ha desarrollado numerosos programas informáticos en código abierto, que se ejecutan desde la terminal y se encuentran disponibles en plataformas como GitHub (https://github.com/) o SourceForge (https://sourceforge.net/). Incluso hay plataformas web como RAST, MG-RAST, EnteroBase o Galaxy (https://usegalaxy.org/), que integran una serie de programas y automatizan el proceso para los usuarios, así como la plataforma PLACNETw para la búsqueda de plásmidos48. Además, los científicos desarrollan pipelines de análisis, que resultan útiles porque agrupan herramientas de software; por ejemplo, en nuestro grupo hemos desarrollado TORMES (https://github.com/nmquijada/tormes 40), un pipeline de código libre para el análisis de genomas bacterianos, que permite ensamblar y anotar el genoma, tipificar el microorganismo por Multilocus Sequence Typing (MLST), obtener los factores de virulencia y de resistencia a los antibióticos, y realizar un análisis pangenómico comparativo de los aislados. Los resultados generados son recopilados en un archivo interactivo en formato web, que puede visualizarse en cualquier navegador, y ser compartido y analizado entre los distintos usuarios de una manera sencilla.
Por su parte, el National Center for Biotechnology Information (NCBI) dispone de un proyecto llamado Reference Sequence (RefSeq), que incluye el Prokaryotic Genome Annotation Pipeline (PGAP), ya en su versión 4.1, y un repositorio de genomas procariotas curados y anotados (www.ncbi.nlm.nih.gov/refseq/)21.
Existen diversas plataformas web con genomas depositados y curados, como EnteroBase3 (https://enterobase.warwick.ac.uk/), donde se pueden analizar y tipificar genomas de enterobacterias por MLST, core genome MLST (cgMLST) o whole genome MLST (wgMLST). Otra interesante plataforma es Pathogenwatch (https://pathogen.watch/), que permite acceder a una serie de análisis automáticos a partir de secuencias propias (ensamblados de novo en formato FASTA) y determinar la especie bacteriana por MLST, así como predecir la resistencia a antibióticos y acceder a secuencias públicas de genomas bacterianos completos (también de hongos y virus), que, en última instancia, pueden integrarse con genomas propios para generar colecciones, que sirven para comparar estas secuencias mediante árboles filogenéticos. Microreact4 (https://microreact.org/showcase) es otra plataforma que permite una visualización integrada de los conjuntos de datos de secuenciación (árboles filogenéticos, determinantes de resistencia y virulencia), datos geográficos y temporales, y otras variables de interés para realizar epidemiología genómica.
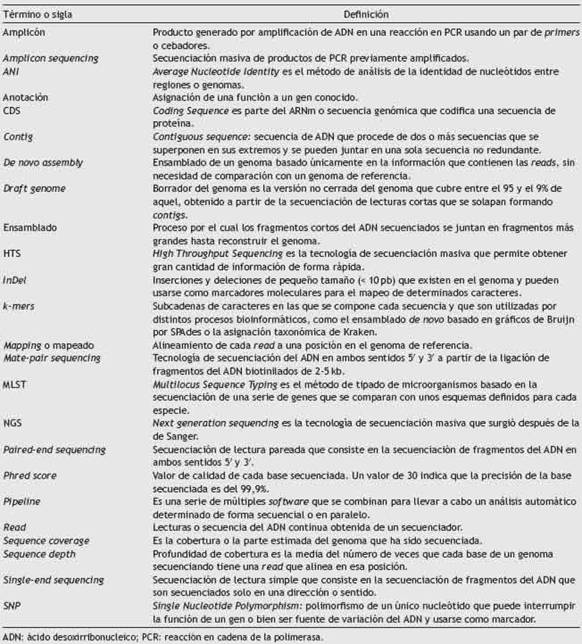
Además del software, es necesario disponer de un hardware o estructura de cómputo de gran capacidad. Consideramos que para poder realizar algunos de los tipos de análisis bioinformáticos, como el mapeo de referencia o el ensamblaje de novo (ambos descritos más adelante), se pueden emplear ordenadores de escritorio o portátiles con software y configuración adecuados (16GB de memoria RAM, 4 núcleos de procesamiento y 1TB de espacio de almacenamiento). Sin embargo, el uso de máquinas de alta gama ubicadas en grandes centros de datos (miles de GB de memoria, hasta 64 núcleos de procesamiento y acceso a cientos o miles de TB de almacenamiento), incluso vinculadas entre sí para construir grupos de cómputo de alto rendimiento o clusters (capaces de analizar simultáneamente cientos o miles de genomas), constituye una práctica eficiente en cuanto al uso de los recursos. Algunos países han creado una infraestructura común para ejecutar los análisis con un gran poder computacional, lo que requiere un conocimiento informático mínimo para el acceso, además de la instalación y el uso de distintos programas o software ya preinstalados en los equipos. Por ejemplo, el Reino Unido creó en 2016 Cloud Infrastructure for Microbial Bioinformatics (CLIMB, www.climb.ac.uk.), un recurso informático para la comunidad científica en microbiología clínica, que provee al investigador de capacidad de procesamiento (múltiples cores y memoria RAM) y memoria de almacenamiento, con numerosos programas preinstalados13. Con el fin de ahorrar al usuario la tarea de instalar programas y entornos, se han creado distintos repositorios, como es la iniciativa Bio-Linux (http://environmentalomics.org/bio-linux/), que permite descargar un sistema operativo (Linux) en el ordenador, con una gran variedad de lenguajes y programas bioinformáticos. Con objeto de iniciar al microbiólogo en la bioinformática y facilitarle su comprensión, en la tabla 2 tabla 2 se presenta un pequeño glosario de terminología básica.
En los apartados subsiguientes se describe la aplicación de la secuenciación masiva en la microbiología clínica; asimismo, se desarrollan en extenso dos de sus principales usos.
Aplicaciones bioinformáticas para la secuenciación masiva en la microbiología clínica
La secuenciación masiva aplicada a la microbiología se puede realizar secuenciando pequeños fragmentos del ácido desoxirribonucleico (ADN) o amplicones previamente amplificados (targeted sequencing), o bien secuenciando todo el ADN previamente fragmentado de forma aleatoria (shotgun sequencing). La aproximación de targeted sequencing permite obtener la secuencia del mismo gen en muchas muestras; por ejemplo, en el caso de genes considerados «relojes moleculares» (como los genes ARNr 16S y 18S), este análisis revela la composición microbiana de cada muestra (bacteriana y fúngica, respectivamente). Mediante shotgun sequencing puede obtenerse el genoma completo de una bacteria. Debido a la corta longitud de las secuencias generadas por algunas plataformas de secuenciación, la obtención de un genoma bacteriano completamente cerrado es una tarea compleja y, en algunos casos, imposible, por lo que las reads se ensamblan en un draft genome o borrador del genoma formado por un número de fragmentos más grandes o contigs, mediante un proceso de ensamblado de las lecturas secuenciadas (de novo o reference-based genome assemblies). El conocimiento del genoma facilita la detección de diferencias (mutaciones, SNPs, InDels) entre genomas (genómica comparada), lo que es de gran utilidad en la identificación de los mecanismos de resistencia a antibióticos y en estudios de epidemiología molecular y dinámica de transmisión, así como de filogenética y filogeografía, y también en la estimación de la velocidad de mutación.
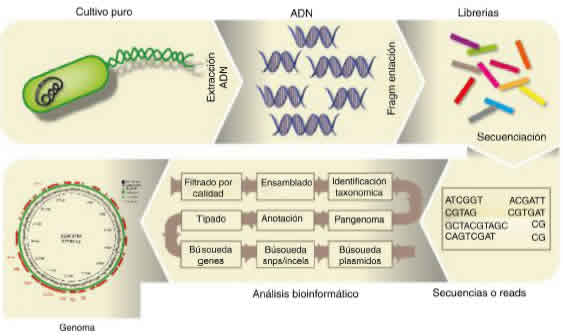
Un experimento de secuenciación masiva consta de 4 etapas principales: la extracción del ADN de la muestra o aislado, la preparación de las bibliotecas o librerías, la secuenciación propiamente dicha y el análisis bioinformático e interpretación de los resultados. Antes de introducir una muestra en el secuenciador, es necesario la preparación de bibliotecas (que denominaremos librerías) de un tamaño determinado, es decir, hay que preparar los fragmentos que van a ser secuenciados. Ello implica fragmentar el ADN por métodos bioquímicos (enzimáticos) o físicos (nebulización o ultrasonido), o bien amplificar fragmentos del ADN (por reacción en cadena de la polimerasa [PCR]). Posteriormente se realiza el marcado de dichos fragmentos con índices (barcoding), para poder secuenciar a la vez múltiples muestras equimolecularmente (multiplexear). También conlleva el reparado de los extremos y la incorporación en los fragmentos del ADN de 2 tipos, un índice que permite la secuenciación de múltiples muestras y un pequeño adaptador del ADN complementario a aquel que existe en el secuenciador, que permite que los fragmentos puedan adherirse a un soporte (flow cell) para ser secuenciados, como es el caso de la tecnología Illumina. La forma de secuenciar puede ser en un sentido de la doble hebra del ADN (single-end sequencing) o en ambos, es decir, 5′ y 3′, en lo que se denomina paired-end sequencing. Este último procedimiento es el más recomendable en microbiología, porque permite obtener fragmentos más largos, de casi 600pb en el caso del MiSeq (Illumina), lo que mejora el ensamblado ulterior.
La etapa de análisis de datos conlleva, a su vez, una serie de pasos: el análisis primario (la generación y el control de calidad), el secundario (alineamiento contra bases de datos específicas, ensamblado de referencia o de novo) y el terciario (generación de datos a partir de los resultados de la etapa de análisis secundario: anotación, búsqueda de SNPs, determinantes de resistencia y/o virulencia, etc.). Una vez concluida la secuenciación, se obtiene un archivo de datos FASTQ para cada uno de los paired-ends (si fue este el tipo de secuenciación realizada), es decir, uno para el forward o secuencia sentido y otro para el reverse o secuencia antisentido correspondientes a cada muestra; dicho archivo contiene las secuencias o reads y los datos de calidad (phred score) basados en código ASCII (en el que cada letra representa un valor numérico), de manera que una secuencia con phred score=10 tiene un 90% de eficacia (una base mal secuenciada cada 10) y una secuencia con phred score=30 tiene un 99,9% de eficacia (una base mal secuenciada cada 1.000). A partir de este archivo FASTQ se realiza en todos los casos el filtrado por calidad, en el que diferentes parámetros como el phred, la longitud, el contenido en GC, los primers, los barcodes, etc., deben ser evaluados con programas como FastQC, en tanto que otros programas como Trimmomatic, Prinseq o Sickle se usan para eliminar restos de barcodes y primers, y filtrar las secuencias basándose en su calidad.
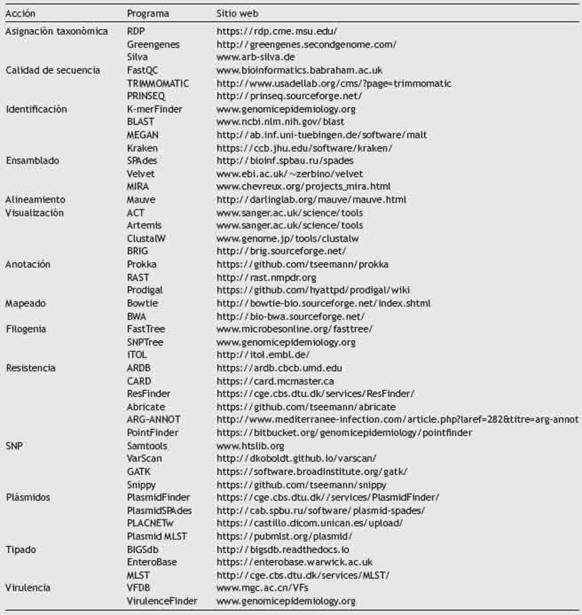
Se han desarrollado numerosos programas; en la tabla 3 tabla 3 se recogen algunos de ellos. Una vez que se obtiene la secuencia filtrada por calidad, se debe analizar mediante alineamiento contra una base de datos específica, para identificar los taxones microbianos de una muestra, o mediante ensamblado y anotado de los genes, para construir el borrador del genoma que permita detectar diferencias entre genomas o identificar los genes de interés, tal y como se va a describir a continuación. En nuestro grupo, hemos realizado trabajos de secuenciación masiva tanto para caracterizar la población microbiana de una muestra con el objeto de identificar marcadores1, como también para asociar la disbiosis intestinal a una enfermedad22. En cuanto a la caracterización de genomas completos, nos ha permitido descubrir nuevos determinantes de resistencia a antibióticos23 y realizar estudios de epidemiología molecular24.
Análisis de la microbiota de una muestra
La identificación y la caracterización de los microorganismos que pueden estar causando una infección o una enfermedad es importante para el tratamiento y la recuperación del paciente, y también para la seguridad del resto de los individuos. Se define como microbiota la composición taxonómica microbiana de una muestra, mientras que el término metagenoma alude al conjunto de genes y genomas de la microbiota. Bajo el término microbioma se engloba el conjunto de genes y, además, sus productos o metabolitos. En todos los casos, se trata de establecer las relaciones ecológicas microbianas en un determinado ambiente o muestra.
Tradicionalmente, la aproximación empleada en estudios de ecología microbiana se basaba en el aislamiento de los microorganismos en cultivo axénico y la purificación de su ADN, o bien la extracción directa del ADN a partir de la muestra y la posterior amplificación por PCR, seguida de la separación de los fragmentos en geles de agarosa o de acrilamida (como sucede en la electroforesis en gel en gradiente desnaturalizante, DGGE) y la identificación de los aislados por secuenciación Sanger. Sin embargo, estos métodos no ofrecen las ventajas de la secuenciación masiva: esta última es independiente del cultivo, se necesita poco ADN de la muestra, tiene suficiente profundidad para identificar microorganismos que estén poco representados y permite multiplexear varias muestras a un bajo coste y en un tiempo reducido.
Como se ha comentado, la mayoría de los métodos de ecología bacteriana se basan en la secuenciación, en concreto, del gen que codifica la subunidad menor del ribosoma 16S (16S ARNr). El tamaño de este gen es de aproximadamente 1.540 pares de bases, dependiendo de la especie bacteriana, y está formado por 9 regiones hipervariables (V1 a V9) y regiones altamente conservadas. Sobre esta base se han definido numerosas combinaciones de primers o cebadores8,29, que permiten amplificar un fragmento del ADN de forma inespecífica en todas las especies bacterianas, pero a su vez es posible realizar la identificación inequívoca de cualquier bacteria, porque la secuenciación de la región interna es característica de cada especie.
La secuenciación Sanger no permitía la mezcla de amplicones ya que el cromatograma no podía ser leído si no era puro, pero en la secuenciación masiva sí, porque esta tecnología individualiza la lectura de cada amplicón. El estudio ecológico molecular de la microbiota mediante NGS consiste en la coamplificación de una región del gen 16S ARN. Por ejemplo, los primers descritos por Klindworth et al.29 amplifican las regiones variables V3-V4 del gen 16S ARNr (~464pb) a partir del ADN extraído directamente de la muestra. Estos fragmentos amplificados, una vez secuenciados, permiten caracterizar la comunidad bacteriana coexistente, con la posibilidad de identificar todas las especies de una muestra; incluso se puede realizar el análisis subespecie de los taxones mediante herramientas como oligotyping16 o la resolución de amplicon sequence variants (ASVs)6. La asignación de filo, clase, orden, familia, género o especie se realiza por comparación con bases de datos como la Ribosomal Database Project (RDP)11, Greengenes37, Silva (www.arb-silva.de), el servidor web del CGE (https://cge.cbs.dtu.dk/services/SpeciesFinder/), o bien realizando un alineamiento empleando la Basic Local Alignment Search Tool (BLAST) de la base de datos del NCBI17. No solo podemos obtener la caracterización de la composición taxonómica, sino también la abundancia relativa de las comunidades microbianas de una muestra. La secuenciación masiva permite el análisis ecológico de la microbiota mediante secuenciación de amplicones, pero también se puede realizar este análisis mediante la secuenciación completa de todos los genomas, es decir, del metagenoma. Esto implica mayor costo y requiere un análisis más complejo y detallado, por lo que esta revisión no profundiza en el análisis metagenómico sensu stricto 41.
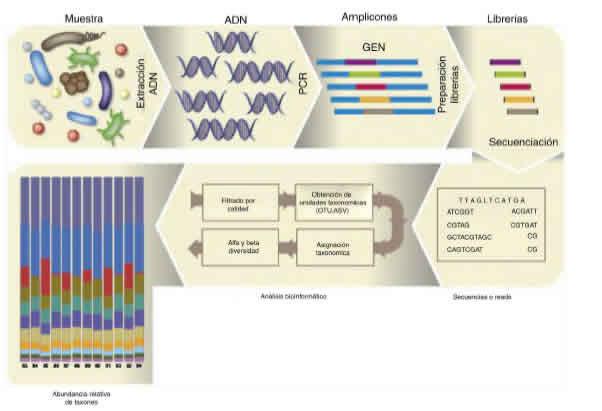
El proceso de análisis de la microbiota de una muestra dada se describe en la figura 1 figura 1. Una vez purificado el ADN microbiano de la muestra, realizada la PCR y la librería, y secuenciados los amplicones, se obtiene el archivo FASTQ. De los datos de este archivo de secuenciación no se obtienen especies sensu stricto, sino que se agrupan las secuencias idénticas con un 97 al 99% de identidad usando el método uclust15 en lo que se denominan operational taxonomic units (OTUs). Sin embargo, hoy ya se utilizan las exact sequence variants (ESVs) ―también denominadas amplicon sequence variants (ASVs)―, que agrupan únicamente aquellas secuencias idénticas6.
Dos herramientas bioinformáticas han popularizado el análisis de la microbiota presente en una muestra: Mothur44 y QIIME7, que recientemente han lanzado su segunda versión. QIIME dispone de tutoriales pormenorizados para ser ejecutados por línea de comando en la terminal y los archivos generados pueden ser visualizados en la web (https://view.qiime2.org/). La última versión introduce el uso del software DADA2, un sistema de filtrado de calidad de secuencia más estricto, que favorece la posterior identificación de ASVs. QIIME2 hace uso de diversos scripts para poder realizar alineamiento de las secuencias, construir gráficos de mapas calientes (heatmaps) de la taxonomía, inferir árboles filogenéticos y efectuar análisis de diversidad alfa (diversidad inherente en una muestra) y de beta diversidad (diversidad entre muestras). La alfa diversidad de una muestra se establece contando el número de especies presentes (richness), la diversidad relativa de las distintas especies (diversity) y la heterogeneidad de la muestra (evenness). Para ello se usan distintas métricas o índices clásicos de ecología: Chao1, Shannon y Simpson. Las curvas de rarefacción representan el número de especies, ASVs, OTUs, etc., versus el número de lecturas, y permiten visualizar de manera sencilla que se ha alcanzado la suficiente profundidad en la secuenciación para identificar aquellos taxones minoritarios en una muestra. La beta diversidad mide la distancia o disimilaridad entre un par de muestras; se pueden realizar distintos análisis estadísticos que representan las muestras en gráficos bi- o tridimensionales (análisis de coordenadas principales, PCoA). Se usan distintas métricas, entre ellas, el índice de Bray-Curtis (método no filogenético que tiene en cuenta la abundancia de OTUs), el unweighted UniFrac (método filogenético que tiene en cuenta la presencia o ausencia de una OTU) y el weighted UniFrac (método filogenético que tiene en cuenta la abundancia de OTUs). Existen muchas otras herramientas estadísticas, cabe destacar entre ellas el LEfSe, un programa gratuito que permite realizar un análisis discriminante lineal efecto-tamaño (LDA effect size) -para identificar las características que explican las diferencias entre clases o predecir marcadores responsables de un fenotipo- y los representa de forma gráfica46 (https://huttenhower.sph.harvard.edu/galaxy/). Pequeñas modificaciones en algunos de los archivos de QIIME o Mothur permiten a LEfSe realizar el análisis estadístico diferencial entre varias muestras que reúnen distintas condiciones y que son objeto de estudio a todos los niveles taxonómicos.
Análisis del genoma bacteriano completo
Para el análisis de secuencias de genomas bacterianos completos, una vez purificado el ADN microbiano de la muestra es preciso someterlo a la fragmentación (física o bioquímica), seguido de la preparación de librerías y la secuenciación, tal como se describe en la figura 2 figura 2. El archivo FASTQ generado por el secuenciador masivo a partir de un genoma bacteriano procedente de un cultivo axénico contiene las secuencias, lecturas o reads, las que, una vez que se han filtrado por su calidad, pueden ser ensambladas en unidades mayores o contigs. Existen varios ensambladores; Loman et al.34 publicaron una revisión al respecto. Cabe distinguir entre los ensambladores que requieren un genoma de referencia (MIRA) y entre los ensambladores de novo (SPAdes). Con secuenciadores como los que comercializa Illumina, con fragmentos secuenciados muy pequeños y habiendo sometido el ADN a una fragmentación enzimática con transposasas (tagmentación) para la preparación de las librerías, resulta imposible cerrar el genoma; sí se podría cerrar usando los secuenciadores con tecnología de molécula única, aunque estos son menos precisos. Por tal razón, ambas tecnologías se consideran complementarias en la actualidad. El resultado, en general, es un draft genome en formato FASTA, formado por cierto número de fragmentos o contigs, que cubren entre el 95 y el 99% del genoma. El número de contigs que forman el genoma es un parámetro que se tiene en cuenta a la hora de evaluar la eficacia del ensamblado, junto con la longitud media y mínima de los contigs, el N50 (longitud del contig más pequeño de aquellos que representan el 50% del genoma), etc. Otro factor a tener en cuenta es la depth o profundidad de cobertura, es decir, el número de veces que una determinada posición nucleotídica fue secuenciada: se consideran genomas de calidad aquellos con una depth superior a 20-25×. Existe también software como QUAST20, que permite analizar la eficiencia del ensamblado, y Qualimap238, que permite analizar la depth de cada posición nucleotídica, más allá de una estimación media.
Posteriormente, los contigs pueden ser visualizados utilizando software como Mauve14o Artemis, y ser ordenados frente a un genoma de referencia utilizando, por ejemplo, Mauve o ACT, que a su vez permiten visualizar los genomas utilizados. La figura 2 ilustra el flujograma del proceso para analizar el genoma bacteriano. El ensamblado es uno de los «cuellos de botella» del proceso, computacionalmente hablando, por lo que existen distintos programas que pueden funcionar directamente desde las reads. Tal es el caso de Kraken, que permite identificar taxonómicamente una especie o género de forma rápida sin ensamblar las reads, utilizando alineamientos exactos de k-mers contra su propia base de datos, por lo que es muy útil para detectar contaminaciones49, o snippy (https://github.com/tseemann/snippy), que mapea las reads contra un genoma de referencia con el objeto de detectar polimorfismos. También el software ARIBA25 permite detectar genes de resistencia a antibióticos, virulencia o replicones plasmídicos a partir de las reads. Existen visualizadores de genomas propiamente dichos, como BLAST Ring Image Generator (BRIG)2, que permiten la comparación visual de varios genomas frente a uno de referencia y localiza los genes anotados.
Una vez que los contigs han sido ordenados en un borrador de genoma más o menos cerrado, se procede a la asignación de las funciones de un genoma, es decir, a su anotación, lo que conlleva una serie de procesos como la búsqueda de posibles genes (coding sequences o CDS) mediante programas como Prodigal26, su traducción a proteínas y la identificación de estas proteínas contra bases de datos (diamond y blastp). Existen herramientas para automatizar el ensamblado en plataformas web, como RAST5, o por línea de comandos en la terminal usando Prokka45. El resultado son distintos archivos con información de anotación del tipo GFF, GBK, GBL, etc., los cuales son siempre dependientes de la base de datos que se haya utilizado para anotar; la del NCBI es una de las más completas. Una de las ventajas de la anotación por programas de línea de comandos radica en la capacidad del usuario de generar bases de datos de genes propias, surgidas a partir de sus investigaciones particulares, ya que, en última instancia, estas mejorarán la calidad de la anotación.
Un objetivo probable cuando se intenta obtener el genoma de una bacteria es poder buscar genes de interés que permitan el genotipado. Por ejemplo, puede ser necesario localizar los genes housekeeping, que permiten obtener el MLST. PubMLST contiene todos los esquemas y, específicamente para Listeria spp., existe el esquema del Instituto Pasteur (http://bigsdb.pasteur.fr/listeria/listeria.html). Asimismo, EnteroBase tiene esquemas para las principales enterobacterias y actualmente también para Clostridioides. Existen plataformas como las MLST32,35o el Plasmid MLST Databases28 y el servidor del Center for Genomic Epidemiology (CGE) (http://www.genomicepidemiology.org/), que permiten obtener el secuenciotipo de un aislado. También en el servidor del CGE se puede serotipificar usando la herramienta SerotypeFinder 2.0, hacer spaTyping en el caso de Staphylococcus aureus, tipificar Escherichia coli por fimbrias con fimTyper, etc. Por otro lado, se pueden buscar genes que codifiquen resistencia o virulencia con software como ARIBA (ya mencionado) o Abricate (https://github.com/tseemann/abricate), que usa la herramienta BLAST del NCBI (http://blast.ncbi.nlm.nih.gov) para buscar en el genoma genes de resistencia a antibióticos que se encuentren en bases de datos como ResFinder (acquired antimicrobial resistance gene finder)50, Comprehensive Antimicrobial Resistance Database (CARD)27, Antibiotic Resistance Genes Database (ARDB) o Antibiotic Resistance Gene-ANNOTation (ARG-ANNOT)19. En cuanto a los genes de virulencia, existe una base de datos denominada Virulence Factor Database (VFDB)10, que contiene los principales determinantes de virulencia bacterianos y puede usarse con la herramienta BLAST.
Una de las aplicaciones más interesantes de la secuenciación masiva es la posibilidad de detectar variantes (SNPs, Indels). Para ello se mapean las reads directamente contra un genoma de referencia utilizando software como SMALT (https://www.sanger.ac.uk/science/tools/smalt-0), Bowtie30o BWA33. El archivo resultante es Sequence Aligmant Map (SAM) y debe ser manipulado con Samtools y VarScan o GATK para generar un archivo Variant Calling File (VCF), que pueda ser considerado como suficiente para definir SNP/InDel. Con esta aproximación se buscan todas aquellas reads que tengan un nucleótido que difiera con el genoma de referencia, es decir, permite obtener todas aquellas reads que soportan un SNP o InDel. No podemos definir un número concreto de reads que pueda ser considerado como suficiente, ya que hay muchos aspectos por considerar (como la calidad de esa posición y otros). Los archivos SAM/BAM y/o VCF pueden visualizarse con Artemis o IGV. Algunos tipos de software como Parsnp permiten la comparación de aislados mediante core-genome-SNP, esto es, mediante la comparación de variantes nucleotídicas en cada una de las posiciones. Teniendo en cuenta la información de los SNP, es posible construir dendrogramas, que pueden ser visualizados con programas como Gingr, FigTree, PhyML o RAxML. El programa FastTree utiliza métodos heurísticos de máxima verosimilitud para generar un árbol a partir de alineamientos de nucleótidos o secuencias proteicas. Los árboles se pueden visualizar y manipular, por ejemplo, con iTOL, con el paquete de R ggtree y con el ya mencionado Figtree.
Otra aplicación de la secuenciación de genomas completos es comparar las regiones del genoma obtenido contra un genoma de referencia. Se pueden comparar ambos genomas utilizando BLAST y visualizarlos mediante Artemis Comparison Tool. Con la información de todos los genes obtenemos el denominado pangenoma (todos los genes presentes en un genoma), el genoma core (genes que están presentes en cualquier genoma dentro de un filotipo) y los genes accesorios (genes que están presentes en un conjunto de aislados dentro del conjunto de datos). Se puede comparar el pangenoma de distintos aislados anotados mediante programas como Roary39, que nos permite identificar a partir del archivo GFF los core genes (genes compartidos en el 99-100% de los genomas comparados), softcore (95-99%), shell (15-95%) y los cloud genes (0-15%), y construir una matriz de distancias para generar un árbol basándose en la presencia/ausencia de genes en las distintas muestras. También existen esquemas de tipificado basados en core genome MLST (cgMLST) y whole genome MLST (wgMLST), es decir, basados en un conjunto fijo de loci conservados en todos los genomas y se usan esquemas específicos de especie32.
Por último, la secuenciación permite obtener tanto la información genética cromosómica como la contenida en plásmidos como material transferible y de replicación autónoma. Así, por ejemplo, el software PlasmidFinder permite la identificación de motivos plasmídicos usando una base de datos no redundante de replicones (secuencias que activan o controlan la replicación del plásmido y son identificativas de un determinado tipo de plásmido) y los asigna a un determinado grupo Inc (grupo de incompatibilidad plasmídica) o Rep9. Posteriormente se siguen distintas aproximaciones para identificar las reads plasmídicas. PlasmidSPAdes es otro software basado en el concepto de que, durante la extracción de ADN, a igual concentración molar de ADN habrá más número de plásmidos que de cromosomas; esta mayor concentración de plásmidos se traduce en un mayor número de reads plasmídicas tras la secuenciación y el programa utiliza un algoritmo que es capaz de separar y tratar de ensamblar estas reads que están en un mayor porcentaje que el supuesto cromosoma. Similarmente, pueden mapearse las reads contra un cromosoma de referencia mediante Bowtie o BWA y efectuar el descarte de aquellas que mapeen (y, por tanto, con posibilidad de ser cromosómicas) y el ensamblado posterior de aquellas que no mapearon con SPAdes. Lanza et al.31 desarrollaron una herramienta denominada PLACNET para la reconstrucción gráfica de plásmidos, cuya versión web48 apareció en 2017.
Cualquiera sea el software, siempre es más interesante realizar los análisis por línea de comandos que en la web, porque se pueden analizar múltiples genomas a la vez y lanzar procesos en paralelo, lo cual reduce el tiempo de procesado (siempre dependiendo de la capacidad de procesamiento del hardware) e independiza al usuario de la disponibilidad de servidores web por lo general más lentos.
Conclusiones y perspectivas futuras
La secuenciación masiva ha transformado la microbiología, sobre todo desde que se han reducido los costes y los tiempos de análisis, gracias al desarrollo de la bioinformática, que va generando nuevos programas de análisis. La masiva generación de datos requerirá cada vez más inversiones en grandes centros de supercomputación, y el desarrollo de programas de código abierto será cada vez más profuso, aunque, paralelamente, se comercializarán paquetes de uso bioinformático que requerirán mínimos conocimientos técnicos.
La secuenciación masiva se impondrá en el análisis microbiano, ya que permite estudiar la epidemiología y trazar microorganismos individuales en pacientes, en los hospitales, en la comunidad y en el planeta en general, además de estudiar su evolución. Por ejemplo, Comas et al.12 han revisado las aportaciones de la secuenciación masiva al diagnóstico y la epidemiología de la tuberculosis y los esfuerzos que se están haciendo para dar el salto a su aplicación en el contexto clínico. Pero la aplicación de la secuenciación masiva va más allá de la medicina humana. Teniendo en cuenta que 7 de cada 10 enfermedades infecciosas son de origen animal, estas tecnologías pueden favorecer la cooperación en el estudio de las enfermedades en humanos y animales, y su interacción en el medio ambiente bajo el concepto «una sola salud»51. Asimismo, también se puede conocer la situación de la comunidad microbiana en el nicho ecológico estudiado (en lugar de obtener información de los microorganismos aislados) y, por tanto, establecer asociaciones microbianas que pueden explicar la etiología o la evolución de algunas enfermedades o síndromes de causa desconocida o no bien definida. La secuenciación masiva, a través de su aplicación a la exploración de la microbiota y del metagenoma, ha cambiado el modo en el que el microbiólogo investiga, y su potencial con las nuevas herramientas bioinformáticas es enorme. El descubrimiento de nuevos genes que codifican resistencias a antibióticos, factores de virulencia, enzimas; en definitiva, de nuevas funciones de la célula procariota, abre un enorme campo para investigar, muy dependiente del avance de los métodos de secuenciación y también del desarrollo de algoritmos informáticos de análisis.
Tanto las bacterias patógenas como las no patógenas pueden representar una amenaza, pero también una oportunidad para la salud. Describir la epidemiología y los mecanismos de virulencia, así como las interacciones con el organismo hospedador, proporciona un conocimiento indispensable para establecer la relación salud-enfermedad y obtener verdaderos triunfos en enfermedades infecciosas y grandes avances en el tratamiento de muchas de ellas. No obstante, los métodos aquí descritos requieren una estandarización y validación para que los resultados sean comparables, como garantía de que la extensa cantidad de datos que se están produciendo son fiables y representan un progreso real en la generación de conocimiento. Además, la secuenciación masiva y su análisis permite la integración de la microbiología humana con la veterinaria, con el objetivo de vigilar los microorganismos zoonóticos conocidos y su transmisión, y de estar atentos a la posible aparición de nuevas amenazas. La comprensión de la salud en términos globales, atendiendo a los microrganismos circulantes entre la población, pero también entre los animales y el medio ambiente, constituye la clave del éxito en el control de las patologías infecciosas presentes y futuras.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.

