










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkLatin American applied research
Print version ISSN 0327-0793
Lat. Am. appl. res. vol.33 no.3 Bahía Blanca July/Sept. 2003
Modeling the electropherogram of small charged molecules in capillary zone electrophoresis
M. V. Piaggio1 and J. A. Deiber2
1 Facultad de Bioquímica y Ciencias Biológicas
Paraje El Pozo - (3000) Santa Fe - Argentina
2 Instituto de Desarrollo Tecnológico para la Industria Química
INTEC (UNL - CONICET)
Güemes 3450 - (3000) Santa Fe - Argentina
treoflu@ceride.gov.ar
Abstract ¾ A model for capillary zone electrophoresis (CZE) is presented to carry out the method development of practical separations involving small charged molecules. The model is based on principles and dynamic equations that contain true physicochemical properties of electrolyte solutions and analytes. The basic variables considered in the model are: injection and detection lengths of analytes, electrical field strength, hydration radius and diffusion coefficient of analytes, pK of analyte terminal groups, pH and ionic strength of electrolyte solution, capillary diameter and length, capillary zeta potential, test temperature and relative mass fractions of analytes. The model is solved numerically to predict the separation of analytes through the resulting electropherogram, which is then compared with experimental data.
Keywords ¾ Capillary Zone Electrophoresis. Electropherogram Modeling. Small Charged Molecules. Hydration Radius. Capillary and Particle Zeta Potentials.
I. INTRODUCTION
Although different techniques of capillary electrophoresis (CE) for analytic purposes have been known from many years ago, their uses have increased and developed substantially during mainly the last decade. At the present time, one can find modern and full automatic apparatuses available commercially allowing separations of analytes in a few minutes. We are concerned in this work with the most commonly used mode of CE designated free-solution capillary electrophoresis and also referred to as capillary zone electrophoresis (CZE).
The basic apparatus of CE (Figure 1) analyzed here is composed of two reservoirs (vials) filled with the background electrolyte (BGE) that are connected through a capillary tube made of fused silica (quartz) the diameter of which is typically in the range of 10 to 100 m m. The sample, which is a mixture of analytes to be separated, is introduced at the inlet of the capillary in a small amount (around 1 to 40 nL) by applying a pressure difference (typically 0.5 psig) during a few seconds (1 to 8 s) after replacing the sample vial for the BGE vial. Although there are other methods for the purposes of introducing the sample, hydrodynamic injection is the most frequently used.

Fig. 1. Basic components of a CE apparatus.
Since the separation principle in CZE is based on the analyte electrophoretic mobilities associated mainly to the effective charge and size of each analyte, electrodes are inserted in the BGE vials to apply a high voltage difference in the range of 5 to 30 kV. Typically the positive electrode or anode is located at the inlet vial and the negative electrode or cathode is at the outlet vial. At a distance from the capillary inlet (detection length) an UV detector is placed to register absorbance (proportional to analyte concentrations in the range of linear response) as function of time. Concentrations of the order of 10-5 to 10-8 M can be detected. Analytes can move along the tube due to both the electroosmotic flow (EOF) and the electrophoretic migration (EM). One should observe that the EOF is the bulk movement of the fluid through the capillary as a consequence of the interaction between the electrical double layer generated by the BGE on the tube wall and the applied electrical field, while the EM is the result of the electrical force on an analyte, considered as a suspended spherical particle. It is then clear that in the context of the experimental framework of CZE, one must choose carefully several operational variables, minimize undesired phenomena like Joule heating and dispersion effects and in general obtain the best experimental conditions for optimal separation (high efficiency and resolution).
In this context of analysis, the interpretation of the electropherogram through electrokinetic theories available in the literature is a practical requirement. In this sense, Reijenga and Kenndler (1994) described the framework of several types of models with different degrees of complexities that can provide these needs. Thus a model of CZE can serve different purposes and depending on them, one may consider a wide variety of phenomena. In relation to this last aspect, Poppe (1998) discussed the concepts of ideal and linear models in CZE, which might be the reference frameworks where the mathematical treatment becomes amenable to analysis without entering in complex numerical calculations (Saville and Palusinski, 1986). Having these aspects in mind, a model can be also useful for performing additional calculations that are necessary in order to arrive at quantitative conclusions.
Therefore, the purpose of this work is to present a non-ideal and linear model of CZE to help one in the method development and optimization of practical separations of small charged molecules (see, for example, Jandik and Bonn, 1993; Khun and Hoffstetter-Khun, 1993; Grossman and Colburn, 1992). Although this model is based on fundamental principles and dynamic equations, it has to be relatively simple to compute and use in the laboratory. The basic variables considered in the model are: injection and detection lengths of analytes, electrical field strength, hydration radius and diffusion coefficient of analytes, pK of analyte terminal groups, pH and ionic strength of electrolyte solution, capillary diameter and length, capillary zeta potential, test temperature and relative mass fractions of analytes.
The numerical code elaborated with the model is able to predict the effect on the electropherogram resolution that yields a change carried out on any fundamental variable listed above. Examples of the prediction capability are presented for different analytes. Experimental data obtained from a commercial equipment of CE are used for comparison with theoretical predictions. In particular we present specific studies involving EOF tracers like dimethyl sulfoxide (DMSO) and caffeine and small charged molecules (theophylline, salicylic acid and benzoic acid).
Before ending this section, it is relevant to indicate that the model proposed here allows one the determination of diffusivity, hydrodynamic radius and zeta potential of analytes, when analyte migration times are available from electropherograms. This aspect has not been fully considered in previous works.
II. MODEL DEVELOPMENT
In this section we present the balance equations of the relevant physicochemical properties that are required to model the electropherogram of CZE. These equations are specialized here in the description of the EOF and EM in the capillary tube characterized by a total length L and radius R. Here the detection length is designated Ld and of course Ld < L. Throughout this work, variables have SI units unless it is stated specifically.
A. Electroomostic Flow in the Capillary
The electroomostic flow is analyzed through the balance equations describing electrokinetic phenomena of charged particles suspended in a fluid (Probstein, 1989; Russell et al., 1991). Thus, mass conservation and balances of momentum, energy and ions are required for the BGE. By considering the electroosmotic flow in the axial direction of the capillary of radius R, the following equations are valid for the steady state flow regime in the cylindrical coordinate system:
 | (1) |
 | (2) |
 | (3) |
In Eqs (1) to (3) g is the gravity acceleration, E is the applied electrical field strength in the tube axial direction z and the divergence of the velocity vector v is considered null. Also, rw is the mixture density evaluated at the wall temperature, m is the viscosity, k is the thermal conductivity and e is the electrical permittivity; all these properties belong to the BGE. Throughout this work e is used for the unit charge. Therefore,  is the distribution of electrical charge per unit volume in the electrolyte solution, which depends on ion valence zi and ion number concentration ni. The condition of neutral charge imposes re = 0 and ni = ni¥ far away from the tube wall. We define the electrical field E = -Ñf, where f is the electrical potential in the electrolyte solution. Since electrostatic laws are valid within the framework of CZE, the electrical field shall be irrotational. The temperature T can vary within the fluid due to Joule effect associated to the term s E × E = sÑf × f where
is the distribution of electrical charge per unit volume in the electrolyte solution, which depends on ion valence zi and ion number concentration ni. The condition of neutral charge imposes re = 0 and ni = ni¥ far away from the tube wall. We define the electrical field E = -Ñf, where f is the electrical potential in the electrolyte solution. Since electrostatic laws are valid within the framework of CZE, the electrical field shall be irrotational. The temperature T can vary within the fluid due to Joule effect associated to the term s E × E = sÑf × f where  is the electrical conductivity and mi is the electrophoretic mobility of ion i. Viscous dissipation is of the order of the water viscosity value and is neglected. In addition, Eq. (3) has associated the balance of ions expressed through ni. In this context of analysis the total diffusion flux Ji# = ni(ui# + vie) of ions i involves two driving forces, one is generated by the Brownian movement and the other is a consequence of the electrical potential. Thus, the ion velocity vi = v + vie + ui# includes the mixture velocity v of N species, the electrophoretic velocity vie and the diffusion velocity ui#. Therefore, the appropriate diffusion constitutive equation is
is the electrical conductivity and mi is the electrophoretic mobility of ion i. Viscous dissipation is of the order of the water viscosity value and is neglected. In addition, Eq. (3) has associated the balance of ions expressed through ni. In this context of analysis the total diffusion flux Ji# = ni(ui# + vie) of ions i involves two driving forces, one is generated by the Brownian movement and the other is a consequence of the electrical potential. Thus, the ion velocity vi = v + vie + ui# includes the mixture velocity v of N species, the electrophoretic velocity vie and the diffusion velocity ui#. Therefore, the appropriate diffusion constitutive equation is  (see, Russel et al., 1989 and Probstein, 1989, for further details).
(see, Russel et al., 1989 and Probstein, 1989, for further details).
Another relevant parameter is the ion hydrodynamic mobility wi = 1/6pm ai expressed as the inverse of the Stokes friction coefficient associated to a spherical particle with hydration radius ai suspended in a flow field.
The following hypothesis are typically found in the description of the electroosmotic flow described by Eqs. (1) to (3): (a) thermal and electrical conductivities, viscosity and electrical permittivity are nearly constant, (b) Boussinesq approximation concerning body forces is valid in relation to the temperature field, and the mixture density is described through r = rw[1 - b(T - Ti)]. In this expression b is the thermal expansion coefficient and sub index w indicates evaluation at the tube wall temperature.
The boundary conditions for the mathematical problem described by Eqs. (1) to (3) consider the non-slip velocity, the capillary zeta potential  and the temperature Tw, all evaluated at the tube wall. Conditions of symmetry at the tube centerline are also imposed. Since the thickness of the electrical double layer near the tube wall is small in relation to the radius of the capillary tube and the electrical potential changes within this region only, the ion number concentration ni can be expressed (Russel et al., 1989)
and the temperature Tw, all evaluated at the tube wall. Conditions of symmetry at the tube centerline are also imposed. Since the thickness of the electrical double layer near the tube wall is small in relation to the radius of the capillary tube and the electrical potential changes within this region only, the ion number concentration ni can be expressed (Russel et al., 1989)  . Having into account these boundary conditions, one obtains for a z-z electrolyte,
. Having into account these boundary conditions, one obtains for a z-z electrolyte,
 | (4) |
 | (5) |
 | (6) |
where  and kB is the Boltzmann constant. We define
and kB is the Boltzmann constant. We define  , which gives an estimation of the electrical double layer thickness lD near the tube wall also designated the Debye length. Here, NA is the Avogadro number and the ionic strength
, which gives an estimation of the electrical double layer thickness lD near the tube wall also designated the Debye length. Here, NA is the Avogadro number and the ionic strength  is expressed in units of Mº mol/L. Since in the CE apparatus, the capillary inlet and outlet enter the BGE and sample vials in the vertical position, the sign (-) stands in Eq. (5) for the case in which the fluid is moving upward in the capillary tube; sign (+) is for the inverse situation. In Eq. (5),
is expressed in units of Mº mol/L. Since in the CE apparatus, the capillary inlet and outlet enter the BGE and sample vials in the vertical position, the sign (-) stands in Eq. (5) for the case in which the fluid is moving upward in the capillary tube; sign (+) is for the inverse situation. In Eq. (5),  is the Grashof number that measures the distortion of the electroosmotic flow due to thermal and electrical effects. This equation shows one that the radius of the fused silica capillary, which is raised to the fourth power, shall be chosen with the criterion of minimizing thermal effects (Piaggio and Deiber, 2000). Therefore, high thermal conductivity and permittivity of BGE and low electrical field strength and ionic strength manifested through the BGE electrical conductivity are also desired to achieve a well shaped electropherogram.
is the Grashof number that measures the distortion of the electroosmotic flow due to thermal and electrical effects. This equation shows one that the radius of the fused silica capillary, which is raised to the fourth power, shall be chosen with the criterion of minimizing thermal effects (Piaggio and Deiber, 2000). Therefore, high thermal conductivity and permittivity of BGE and low electrical field strength and ionic strength manifested through the BGE electrical conductivity are also desired to achieve a well shaped electropherogram.
Equation (5) indicates that significant distortions of the electroosmotic flow when the tube diameter D=2R is high can be obtained due to the enhancement of thermal and electrical effects. In fact, since < 0 for fused silica capillary, it is observed that the upward EOF for high NGr can be different from the desired near plug flow. The direct consequence of the radial varying flow is the increment of the undesired axial Taylor - Aris dispersion phenomenon with the loss of resolution in the separation of analytes. In regards to the downward EOF for high NGr this situation becomes even more critical in the sense that the velocity profile can be directionally inverted near the tube centerline generating recirculating zones within the capillary. This phenomenon is responsible for the catastrophic failure frequently found in the CZE experiments. Calculations show in general that the capillary diameter shall not exceed a value of around 150 m m when the applied voltage difference is around 30 kV.
The theory presented in this section should then be used to guarantee that the electroosmotic velocity v is consistent with the following expression when NGr is small (see Eq. (5) and Russel et al., 1989, for further details):
 | (7) |
where the variation of the electrical potential is also neglected within the small region R - lD £r£R because lD is very small in practice. Eq. (7) will be used throughout this work having into account that CZE variables are chosen so that the Grashof number is appropriately minimized according to the suggestions provided in this section.
B. Injection of Analytes
In this model the injection driven by pressure is considered. For the inception of flow under a sudden applied pressure gradient Dp / L, the volume V of sample injected during a time t in the tube of length L satisfies the following expression, which is obtained from the balance of momentum of the fluid within the capillary:
 | (8) |
In Eq. (8), teff is the effective time of injection accounting for the transient effects associated to the fact that the sample starts from rest before moving into the capillary tube. In most of the practical circumstances teff » t and the evaluation of Eq. (8) to obtain V is carried out directly with the injection time t. In the modeling of CZE the amount of sample introduced in the capillary inlet is considered ideally as a near plug with a length h defined through the volume of sample given by Eq. (8), which satisfies h = V /(p R2).
C. Capillary Zeta Potential
An important conclusion in the literature concerning the modeling of CZE is that a z-z electrolyte solution is neutral at the centerline of the capillary tube when the Debye length lD satisfies R / lD ³ 10 (Gross and Osterle, 1968). In CZE this relation is fully achieved and for the purposes of calculating the capillary zeta potential, the hypothesis of flat surface is valid. Thus, curvature of the tube can be neglected. Since Eq. (6) is deduced under these considerations, the specific charge of the capillary surface is readily obtained from this equation as  . Therefore, to evaluate the specific charge qt should be modeled (see, for example, Healy and White, 1978) from basic information concerning: a) the pK of silanol groups attached to the external layer of the fused silica wall and, b) the total number of acid groups per unit area. This subject is under research at the present time and there is not a unique value of the effective pK of silanol groups available in the literature (Jandik and Bonn, 1993). This context of analysis indicates that further research is required to elucidate better the interaction between electrolyte solution and effective charge at the tube wall for different pH and I.
. Therefore, to evaluate the specific charge qt should be modeled (see, for example, Healy and White, 1978) from basic information concerning: a) the pK of silanol groups attached to the external layer of the fused silica wall and, b) the total number of acid groups per unit area. This subject is under research at the present time and there is not a unique value of the effective pK of silanol groups available in the literature (Jandik and Bonn, 1993). This context of analysis indicates that further research is required to elucidate better the interaction between electrolyte solution and effective charge at the tube wall for different pH and I.
Therefore, we decided to evaluate experimentally by using two neutral markers: dimethyl sulfoxide (DMSO) and caffeine. Injection times of one and three seconds were used, respectively, to introduce these markers into the capillary with a diameter of 50 m m and the migration times tm of markers were measured at different pH and I for E = 15 kV. Since markers are neutral, they move along the capillary tube only due to the EOF. Thus from Eq. (7) one readily obtains,
 | (9) |
where the electroosmotic velocity is considered a near plug flow, as one should expect when the independent variables have been chosen for a Grashof number minimized, as explained in a previous section above. The capillary zeta potential can be evaluated with Eq. (9) when data of tm are available as shown in Table 1.
Table 1. Capillary zeta potential obtained from Eq. (9) for different pH and ionic strength I.

D. Electrophoretic Migration
The modeling of a charged particle migration in an electrolyte solution can be carried out with the same basic equations used to describe the electroosmotic flow. Here, the electrolyte solution develops a velocity profile around the particle and specific boundary conditions are satisfied on the particle-fluid interface. Thus the fluid velocity v, the electrical potential f and the diffusion flux Ji# fulfill, v = vie, (Ji# - nivie) × n = 0 and (eÑf)×n = qi on the surface of the particle i with a specific charge qi. Here, nis the unit vector perpendicular to the particle surface. Also, v ® 0, ni ® ni¥ and -Ñf ® E far away from this particle. The problem thus generated is rather complex to solve. Nevertheless, several conditions proper to CZE facilitate the analysis of the electrophoretic particle migration superposed to the electroosmotic flow in the tube. Fortunately the particle Reynolds number is of the order of 10-4 and inertial effects in the flow field can be neglected. On the other hand, charge convection cannot be discarded in general, because the Peclet number is around 1/2 (Russel et al., 1989). Although the most relevant expressions that allow one to estimate the asymptotic electrophoretic velocities are known, in a more general context, the consideration of numerical solutions reported in the literature (O´Brien and White, 1978) suggests to one that the relation between electrophoretic velocity vie and particle zeta potential  may present complex responses for high , like the saturation of the migration velocity (evolution toward a constant value for increasing particle zeta potentials) as well as the presence of a maximum in the relation between particle mobility and particle zeta potential. In particular, for the small charged molecules (aik < 1) studied in this work, Henry ´s solution is valid and can be expressed (see, for instance, Probstein, 1989 and Russel et al.,1989),
may present complex responses for high , like the saturation of the migration velocity (evolution toward a constant value for increasing particle zeta potentials) as well as the presence of a maximum in the relation between particle mobility and particle zeta potential. In particular, for the small charged molecules (aik < 1) studied in this work, Henry ´s solution is valid and can be expressed (see, for instance, Probstein, 1989 and Russel et al.,1989),
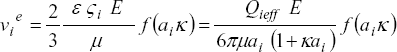 | (10) |
where the effective charge  is proportional to the charge fraction a± calculated from the dissociation constants of base and acid groups of analytes as indicated below. For practical reasons, in our calculations Henry ´s function is represented through f(aik) = 0.988 + 0.509 /{1 + (8.416 / aik)0.985}, which fits well the corresponding numerical data. Therefore, for aik < 1 Eq. (10) can be directly considered in the CZE model formulated here.
is proportional to the charge fraction a± calculated from the dissociation constants of base and acid groups of analytes as indicated below. For practical reasons, in our calculations Henry ´s function is represented through f(aik) = 0.988 + 0.509 /{1 + (8.416 / aik)0.985}, which fits well the corresponding numerical data. Therefore, for aik < 1 Eq. (10) can be directly considered in the CZE model formulated here.
E. Axial Detection of Analytes
The theory involving the axial detection of analytes is a crucial theoretical problem defining the quality of the CZE model. As it will become evident in this section, in the detection of analytes one has to estimate first the effective charge of particle required in Eq. (10) according to the dissociation constants for acid and base groups of analytes (see, for example, Poppe 1998, for a critical discussion of this specific aspect, and also Kenndler, 1998). Thus one can define, for example, the negative charge fraction ai- of an acid group with a given pKa through the expression 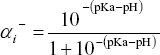 . Other expressions can be found for the positive and negative charge fractions of basic groups.
. Other expressions can be found for the positive and negative charge fractions of basic groups.
Once the effective charge of analytes, the capillary zeta potential and the EOF of the BGE have been estimated with the theoretical framework of the previous sections for well specified operational conditions of the CZE run, the migrations of particles (analytes) must be evaluated along the axial coordinate z of the capillary tube in order to register at the UV detector the absorbance as function of time. Then from the absorbance peaks (proportional to concentration peaks) the migration times of analytes are obtained as far as the electropherogram resolution is of course appropriate. By considering the analysis of Poppe (1998) concerning the classification of models according to the types of phenomena included in the balance of species, it is clear that the CZE model proposed here may be considered non-ideal and linear in the sense that dispersion effects are relevant and analyte samples are introduced in the capillary tube under diluted conditions.
With these physical considerations and when the adsorption of analytes on the capillary wall is not present, the balance involving the ion number concentration in cylindrical coordinates to describe the movement of charged species along the capillary tube toward the UV detector is:
 | (11) |
where Ci is the molar concentration of analyte i and Di » kBT / 6p m ai is the Stokes-Einstein molecular diffusion coefficient. It is important to point out here that Di has associated an unique value of ai for the case of strong electrolytes only, while for a weak electrolyte one should interpret the hydration radius as an average value calculated with the radii of neutral and charged fractions (see, for example, Saville and Palusinski, 1986). Eq. (11) poses the classical Taylor - Aris axial diffusion problem, which allows one to solve the radial average concentration <Ci> in terms of position z, time t and the z-components of analyte velocities (v + vie). In this sense, the Taylor-Aris effective diffusion coefficient is Dieff = kBT(1 + cPe2) / (6p mai), where c depends on the effective velocity profile placed in Eq. (11). Here the Peclet number Pei = (v + vie)ai / Di shall be less than 0.4L / R for the expression giving Dieff to be valid; this condition is readily satisfied with the characteristic scales of CZE. Although the experimental CZE of small charged analytes yields typically Pei £ 100, one should observe that axial dispersion might be enhanced mainly for high applied electrical field and capillary radius; i.e., for high Grashof number. Otherwise, for the case of near plug flow it is well known that c » 0.
Although Taylor-Aris axial diffusion may be negligible because a near plug flow is generally achieved for small orders of NGr (see Eq. (7)), one must account also several other dispersion phenomena in the total effective dispersion coefficient DTieff » Di(1 +  ), which includes all the dispersion effects present in CZE through the analyte dispersion parameter . This parameter can be interpreted as the sum of ratios of each dispersion effect to the molecular diffusion coefficient Di. Therefore, one can define
), which includes all the dispersion effects present in CZE through the analyte dispersion parameter . This parameter can be interpreted as the sum of ratios of each dispersion effect to the molecular diffusion coefficient Di. Therefore, one can define  where tmi is the migration time of analyte i and the most relevant theoretical plate heights Hj are (Kenndler, 1998): R2 <v> /(48Di) for hydrodynamic analyte injection at the average velocity <v> achieved with an applied pressure difference, Ap2 /(12Ld) for aperture Ap of the UV detector,
where tmi is the migration time of analyte i and the most relevant theoretical plate heights Hj are (Kenndler, 1998): R2 <v> /(48Di) for hydrodynamic analyte injection at the average velocity <v> achieved with an applied pressure difference, Ap2 /(12Ld) for aperture Ap of the UV detector,  for thermal dispersion, 2Di /(E mi)µ c for the effective axial dispersion already discussed above, 4coCih / 9 for concentration overload (neglected due to sample dilution) where co is an empirical parameter, [K2 R /(R + 2K)/Di + 4K /(R + 2K)/ Kd]<v> for wall adsorption where Kd is the rate constant of desorption and K is the distribution coefficient of wall adsorption, and R2 Ld /(4rc2) for the capillary coiling with radius rc associated to the conexion between upward and downward capillary branches. Table 2 reports estimated values of by using the above equations.
for thermal dispersion, 2Di /(E mi)µ c for the effective axial dispersion already discussed above, 4coCih / 9 for concentration overload (neglected due to sample dilution) where co is an empirical parameter, [K2 R /(R + 2K)/Di + 4K /(R + 2K)/ Kd]<v> for wall adsorption where Kd is the rate constant of desorption and K is the distribution coefficient of wall adsorption, and R2 Ld /(4rc2) for the capillary coiling with radius rc associated to the conexion between upward and downward capillary branches. Table 2 reports estimated values of by using the above equations.
With these considerations and from the radial average of Eq. (11), one obtains:
 | (12) |
Eq. (12) requires appropriate initial and boundary conditions to obtain relievable concentration profiles as function of detection time. In general one must consider that the injection time of sample affects the solution obtained for <Ci> through the length h of the sample volume assumed as a near plug. Therefore, since the initial condition for Eq. (12) requires that the amount of sample introduced initially at the tube inlet be confined in a finite length, one imposes,
<Ci>=<Ci>o, t = 0, -h / 2 £ z £ h / 2 (13)
when the origin of the coordinate system is placed at the center of the idealized sample plug. <Ci>o is the analyte concentration in the sample vial. During the CZE run, the boundary conditions to be satisfied are:
<Ci>=0, t > 0, - ¥ < z < ¥ (14)
Solutions of Eq. (12) for finite h and h ® 0 can be found in the literature (Crank, 1976).
It is then clear that Eqs. (1) to (14) define fully the non-ideal and linear model of CZE proposed here to determine the appropriate conditions for CZE separation of a mixture of analytes. Therefore, the superposition of solutions obtained for <Ci> as function of time t, and hence migration times tmi, gives one the electropherogram of N analytes. For this purpose a simple numerical code was elaborated with these equations to predict the electropherogram of small charged molecules. Examples of the prediction capability of the model are presented for three analytes: benzoic and salicylic acids and theophylline.
III. EXPERIMENTAL TECHNIQUE
A P-ACE 5010 Beckman Instrument with UV detector was used for the electropherograms involving EOF markers and analytes (DSMO, caffeine, theophylline, benzoic acid, salicylic acid). Standard solutions of analytes were prepared with deionized water at a concentration of 250 m g/ml. The detector was placed at 0.20 m from the inlet of the fussed silica capillary (L= 0.27 m and D=50 m m). The applied voltage was 15 kV and the temperature was fixed at 25 ° C. Hydrodynamic injection of analytes was performed at a pressure difference of 0.5 psig with an injection time of 3 s for standard runs. The buffer was borate at different pH and I (see Table 1). Solutions were prepared by dissolving the appropriate mass of boric acid in distilled water at a concentration of 0.1 M. Then, the pH was adjusted with NaOH 10 M before a small dilution to obtain the final volume. The values if ionic strength obtained were in the range from 10 to 90 mM.
IV. PREDICTION OF ELECTROPHEROGRAMS AND DISCUSSION
To validate the model of CZE, numerical predictions are fitted to experimental data of each analyte migration time, for different pH and I. These data are obtained from the available commercial equipment.
Table 2. Experimental migration time tmi of analytes for different pH and ionic strength I, and numerical estimation of physicochemical properties.

Therefore, with this strategy involving the interplay between theory and experiments, the estimation of hydration radius, diffusion coefficient and zeta potential of each analytes are obtained and reported in Table 2. The values of these properties have the expected order of magnitude (see also Jandik and Bonn, 1993) indicating that the model is providing a good prediction of each analyte electropherogram involving the relatively small molecules considered here.
By increasing pH and I, one observes in Table 2 that ai decreases and - increases consistently with the migration times of analytes. In fact, for higher negative values of particle zeta potentials the effective velocities of particles become lower since, in our experimental setup, the detection of analytes is placed near the negative electrode, while the injection of sample is carried out on the side of the positive electrode (Figure 1). Consequently the electrophoretic migration is against the electroosmotic flow for negatively charged particles. While benzoic and salicylic acids are fully ionized in the pH range worked here, one should observe that theophylline is changing the effective charge fraction ai- (Table 2) from near zero at pH » 8 to -0.94 at pH » 10 as a consequence of the dissociation constant with pK = 8.77. In this sense a transition of the hydration radius at around pH » 9 is consistently observed in Table 2.
On the base of Table 2, Figure 2 illustrates how different values of pH and I can affect the resolution of electropherograms involving the separation of theophylline and salicylic and benzoic acids, which is the main target of this work. Thus, by using the data of Table 2 concerning each analyte, we find that a good resolution is obtained for pH » 10 and I » 0.0882. This theoretical prediction was also validated by the experimental electropherogram indicating that the condition of linear model is fully satisfied for the cases studied here. Furthermore, at pH » 9 and I » 0.0439 the peaks of benzoic and salicylic acids are fully superposed and the separation is not possible for these values of the BGE. Thus, we have a clear indication of the optimal physicochemical conditions for analytes separation from the proposed model as illustrated in Figure 2. In addition, it is observed that at pH » 8.5 the peaks of the salicylic and benzoic acids captured by the UV detector are inverted.
V. CONCLUSIONS
Apart from being a widely used analytical tool, this work suggests that CZE has a high potential for estimating physicochemical properties like hydration radius, diffusion coefficient and zeta potential of analytes, when the electropherogram is modeled and interpreted through the balance equations of transport phenomena coupled to electrokinetic effects. More specifically: 1) A model of capillary zone electrophoresis (non-ideal and linear) involving small charged molecules is validated with experimental data. 2) This model provides quantitative predictions of practical interest for the electropherogram interpretation of a mixture of analytes.



Fig. 2. Model prediction of electropherograms (dimensionless concentrations versus time) for the separation of theophylline (T), benzoic (BA) and salicylic (SA) acids for different pH and ionic strength I (Table 2) of the BGE.
VI. ASPECTS FOR FURTHER RESEARCHES
In the framework of the model proposed here, further research is still required to elucidate better the interaction between the BGE and the effective charge of tube wall. One also requires to investigate particular situations involving the non-ideal and non-linear model of CZE for practical applications. This type of model has been studied numerically in the literature by introducing a perturbation in the basic mathematical problem through the small parameter ekBT /(4h2e2 | zi | ni¥) (see, for example, Saville and Palusinski, 1986).
Acknowledgments
The authors are thankful for financial aid received from the Secretaría de Ciencia y Técnica de la UNIVERSIDAD NACIONAL del LITORAL - Programación CAI+D:96-Proyecto Nro. 112.
REFERENCES
1. Crank, J., The Mathematics of Diffusion. Claredon Press, Oxford (1976). [ Links ]
2. Gross, R.J. and J.F. Osterle, "Membrane Transport Characteristics of Ultrafine Capillaries", J. of Chemical Physics, 49, 228-234 (1968). [ Links ]
3. Grossman, P.D and J. C. Colburn (Eds.), Capillary Electrophoresis: Theory and Practice. Academic Press, USA (1992). [ Links ]
4. Healy T.W and L.R. White, "Ionizable Surface Group Models of Aqueous Interfaces", Advances in Colloid and Interface Science, 9, 303-345 (1978). [ Links ]
5. Jandik, P. and G. Bonn, Capillary Electrophoresis of Small Molecules and Ions. VCH Publishers. New York, US (1993). [ Links ]
6. Kenndler, E., Theory of Capillary Zone Electrophoresis, in "High Performance Capillary Electophoresis", Khaledi, M.G (Ed.). Chemical Analysis Series, 146, Chapter 2, John Wiley & Sons, Inc (1998). [ Links ]
7. Khun, R. and S. Hoffstetter-Khun, Capillary Electrophoresis: Principles and Practice. Springer-Verlag. Cambridge, UK (1993). [ Links ]
8. O'Brien, R.W. and L.R. White, "Electrophoretic Mobility of a Spherical Colloidal Particle", J. Chem. Soc. Faraday Trans., 74, 1607-1626 (1978). [ Links ]
9. Piaggio, M. V. and J. A. Deiber, "A Simple Theoretical Model for Capillary Zone Electrophoresis Method Development". In Proceedings of 6th Latin American Symposium of Biotechnology, Biomedical, Biopharmaceutical and Industrial Applications of Capillary Electrophoresis and Microchip Technology. Punta del Este, Uruguay, 57 (2000). [ Links ]
10. Poppe, H., "Theory of Capillary Zone Electrophoresis", Advances in Chromatography, 38, 233-300 (1998). [ Links ]
11. Probstein, R. F., Physicochemical Hydrodynamics. Butterworths. New York, US (1989). [ Links ]
12. Reijenga, J. C. and E. Kenndler, "Computational simulation of migration and dispersion in free capillary zone electrophoresis. I. Description of the theoretical model", J. Chromatography A, 659, 403-415 (1994). [ Links ]
13. Russel, W. B., D. A. Saville and W. R. Schowalter, Colloidal Dispersions. Cambridge University Press. Cambridge, UK (1989). [ Links ]
14. Saville, D. A. and O. A. Palusinski, "Theory of Electrophoretic Separations. Part I: Formulation of a Mathematical Model", AIChE Journal, 32, 207-214 (1986). [ Links ]

