










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkLatin American applied research
versión impresa ISSN 0327-0793
Lat. Am. appl. res. v.33 n.3 Bahía Blanca jul./sept. 2003
Catalytic abatement jof pollutants from diesel exhausts
M. L. Pisarello, V. G. Milt , M. A. Peralta, C. A. Querini and E. E. Miró
Instituto de Investigaciones en Catálisis y Petroquímica - INCAPE (FIQ, UNL-CONICET)
Santiago del Estero 2654 , 3000 Santa Fe, Argentina
querini@fiqus.unl.edu.ar
Abstract ¾ K and Co,K supported catalysts were studied to determine the mechanism of the soot combustion. The thermal stability of these catalysts and the resistance to water and sulfur are also addressed. The support plays an important role. La2O3 contributes through the formation of carbonate-type intermediates and CeO2 supplies the oxygen necessary for the redox mechanism to take place. However, the addition of Co to the catalyst supported on La2O3, to supply the redox function, leads to a loss of the thermal stability as a consequence of the formation of a perovskite structure. The soot-catalyst contacting phenomenon was also addressed. A synergic La-K effect was observed, by which mechanical mixtures of soot with K-La2O3 showed higher rates than those observed when K and La were deposited on soot. Preliminary experiments using barium as a catalytic trap for NOx and the simultaneous removal of NOx and soot are also presented.
Keywords ¾ Soot Combustion. Nitric Oxide Removal. Cobalt – Potassium Catalysts.
I. INTRODUCTION
An active search has been under way in the last several years to find active catalysts for the abatement of diesel exhaust contaminants. The main pollutants emitted for this type of engines are diesel soot particles and nitrogen oxides. The combination of traps and oxidation catalysts appears to be the most plausible after-treatment technique to eliminate soot particles (Heck and Farrauto, 1995). The catalysts to be used in the combustion of diesel soot must be stable to high temperatures and the presence of other combustion products. On the other hand, the activity should be high enough at temperatures as low as 200° C for low charge diesel engines, unless an external heating power is supplied to the catalytic filter. In any case, the catalyst stability is a key factor in determining its applicability in a commercial scale.
Studies with a large number of formulations have been reported during the last few years, and the soot-catalyst contact appears to be one of the most important problems to be overcome (van Setteen et al., 2000). We have previously studied the catalytic behavior of catalysts containing cobalt and/or potassium supported on La2O3, CeO2, and MgO, mixed with soot under tight contact conditions (Miró et al, 1999, Querini et al, 1998, Querini et al, 1999). Based on these results and results presented in this work, the reaction mechanisms are proposed, being slightly different for each support. In this work, Barium has been included in the formulation of the more active catalysts, since it has been used as a NOx trap (Mahzoul et al., 1999). The activity and stability of these catalysts is therefore also addressed.
II. EXPERIMENTAL
A. Soot and Catalyst preparation
The soot was obtained by burning commercial diesel fuel (Repsol -YPF, Argentina) as described elsewhere (Querini et al, 1998). Catalysts were prepared by wet impregnation, with different amounts of potassium (0, 4,5 and 7,5 wt.%), cobalt (0 and 12 wt.%), and/or barium (10, 16, 22 wt.%) by using KOH, Co(NO3)2 and Ba(CH3COO)2 respectively, as starting materials. Cerium oxide and lanthanum oxide were used as support. The support was added to a suspension of the solution containing K, Co, and/or Ba, which was stirred at 90° C until a paste was formed. Then, the mixture was dried at 100° C and calcined at different temperatures (400 – 700oC).
To investigate about the soot-catalyst contacting effects, samples of soot directly impregnated with either (K) or (K, La) were prepared. The impregnation procedure was carried out from a slurry of soot and solutions of the salts (Lanthanum acetate, Potassium hydroxide) in methanol. Typically, 5 wt.% of the metals were loaded and the paste formed after stirring at 30oC was dried at 100oC.
B. Activity test
The catalytic activity for the combustion of soot was determined by temperature-programmed-oxidation (TPO), of carefully prepared mixtures of catalyst and soot (20:1). The catalysts were mixed with the soot in a mortar, during 3-5 min. A gaseous flow with 6% oxygen in nitrogen was used and the temperature was increased at a rate of 12° C/min, using 10 mg of the mechanical mixture. Experiments using NO + O2 as feed were also carried out. Stability tests were also performed, treating the catalyst with 1000 ppm SO2 at 400° C, and with air that was saturated with water at 25° C, also at 400° C.
C. Catalyst Characterization
The High Frequency CO2 Pulses technique was carried out at 500, 400, and 25° C by sending pulses of 0.135 m mol of CO2 in He carrier every 10 sec. Thermal treatments between pulse cycles were also carried out. Typically, the catalyst was heated up to 700° C, and cooled down to 500° C where a new cycle of pulses was carried out. The CO2 is detected with an FID after methanation, as described elsewhere (Pissarello et al., 2002). The XPS spectra were obtained at room temperature with a computer-controlled Shimadzu ESCA 750 instrument, using MgKa radiation. Microbalance studies were performed in a Cahn 2000 equipment using a mixture of NO (3.75%) + O2 (18.35%) (He balance). When a constant weight value was obtained at 70oC, the sample was heated up to 490oC at 5oC/min, maintained at this temperature for 10 minutes and cooled down to 70oC. At this temperature, the feeding mixture was changed to He and the procedure was repeated.
III. RESULTS AND DISCUSSION
A. Soot combustion experiments
Catalysts with K supported on lanthanum oxides present good activity with a maximum in the TPO profile between 350 and 400ºC (Fig. 1A). K(4,5%), Co-K(4,5%), and Co-K(7,5%) catalysts calcined at 400ºC present the same activity which is higher than that of Co. When the calcination temperature is increased at 700ºC, activity decreases in all Co-containing catalysts, producing a shift in the TPO maximum of about 20-30ºC (Fig. 1B). No activity decrease is found in the case of K/La2O3 catalysts. This increase in the calcination temperature produces a decrease in the amount of Co3O4, indicated by a decrease of H2 consumption in the TPR profile. The increase in the calcination temperature favors the formation of a perovskite-type mixed oxide (LaCoO3) (observed by XRD) (Miró et al., 2000) which is not reducible at temperatures lower than 700oC. The increase of the K content also favors the formation of the perovskite due to its melting effect. The higher the amount of K , the higher the amount of LaCoO3 that is detected by XRD.
On the other hand, XPS results shown in Table 1 indicate that those catalysts shown in Fig. 1 A, the one that presents the best activity (K(4.5)/La2O3), shows the highest value for the surface K/La ratio. K(4,5)/La2O3 doest not present reducible species observable by TPR.
Table 1. K/La and O/La atomic surface ratios measured by XPS


Fig. 1. TPO experiments with different catalysts and pretreatment. Support: La2O3 Gaseous feed: O2 (6%) diluted in He.
In previous works (Querini et al., 1998, Querini et al 1999) we found that a correlation between the amount of Co3O4 and the activity for soot combustion when the support is MgO there exists, which indicates that in the reaction mechanism the redox property of the catalysts is of central importance. However, the MgO support fulfills no function, the presence of Co and K being necessary. Co brings the redox property and K favors the formation of intermediate reaction carbonates. In the case of CeO2 , the presence of an oxide with redox capacity (Co3O4) does not result in any significant increase of activity (Miró et al., 1999), since the support is responsible for practically all the redox capacity. Very similar results are observed both for K/CeO2 and for Co,K/CeO2, which present good activity by burning soot around 300ºC. Neither in CeO2 nor in La2O3 is any correlation observed between activity and reducibility measured by TPR, which does occur in the case of MgO. In those materials, the support provides an alternative route for soot abatement.
B. Interaction of catalysts with CO2. Carbonate species as reaction intermediates.
In order to evaluate the interaction of catalysts with CO2, which is the main product of the reaction, CO2 pulses were injected at different temperatures. It was observed (Querini et al, 1998, Querini et al, 1999) that not only catalysts containing potassium interact with CO2, but also La2O3, producing a distortion in the CO2 peak. This takes place at temperatures close to those of reaction, between 400 and 500ºC, which indicates a strong interaction of CO2 with these components, and a subsequent desorption. The addition of K to the Co catalysts improves the interaction with CO2, which is observed as a smaller CO2 peak at the reactor outlet. To a great extent this interaction is due to La2O3 (which does not occur in the other supports under study) which is supplying a reaction path similar to that supplied by K in MgO or CeO2, with the formation of an intermediate carbonate-type compound. This compound consumes the C from the soot, and decomposes releasing the CO2. Neither MgO nor CeO2 without K show interaction with the CO2, while La2O3 displays a high level of interaction.
For this reason, the K/La2O3 catalyst is very active, and does not require the presence of Co3O4, which supplies the redox property. Likewise, that is why no direct correlation between reducibility and activity is observed.
Table 2. Contribution of each component to catalyst activity
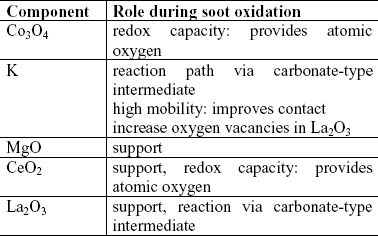
Table 2 summarizes the reaction mechanisms of each one of the supports under study. The oxygen supply for the reaction is also an important stage in the mechanism. In the case of CeO2, the support supplies this oxygen through the redox mechanism (Miró et al., 1999). In Co catalysts, the oxide of this metal is the one which provides the oxygen. The formation of intermediate carbonate-type compounds seems to be very important in the reaction mechanism, as previously reported elsewhere (Querini et al, 1998; Querini et al, 1999).

Fig. 2. High Frequency CO2 Pulses with La2O3 support, at 25 oC, 400oC, and 500oC; (1)first cycle; (2) second cycle after heating at 700oC. Single CO2 pulse at 25 oC, 400oC, and 500oC.
Figure 2 shows results of the CO2 pulses on pure La2O3 both at high and at low frequency. The CO2 peak comes out of the cell with a distortion that depends on the interaction with the catalyst. If the interaction is too high, no CO2 is detected at the cell outlet. If there is no interaction, the peak is sharp. This is clearly observed when a single CO2 pulse is sent to the catalyst cell (low frequency pulse). At 25oC, there is no interaction between the CO2 and the catalyst, correspondingly, the peak comes out very sharp and with the same height as the blank. At 400oC and 500oC, the interaction is high enough to highly distort the shape of the peak. When several pulses are consecutively sent to the cell, the system might reach a pseudo steady state, with a constant amplitude of oscillation, as long as the adsorption – desorption process is fast enough. If the amplitude is lower than the blank, without catalyst or with a catalyst at a temperature where there is no interaction, it means that the dynamics of the process allows the surface to adsorb – desorb the CO2 at the same average rate as the injection. In fact, in the present study with La2O3 support , what we are calling ´adsorption – desorption´, corresponds to the formation of carbonates and its decomposition.
The curve shown in Fig. 2 obtained at 25° C, corresponds to the blank, since at this temperature there is no interaction between the lanthanum oxide and the CO2. The first adsorption at 500° C indicates that there is a strong interaction between the CO2 and the support, and it does not reach the pseudo-steady state with a constant amplitude during the first 20 pulses of the experiment, although it is apparent that it is approaching that state.
After heating at 700° C, the interaction decreases (curve 2, 500° C), being it possible for the catalyst to reach a steady oscillation, i.e., the CO2 is adsorbed and desorbed at the same average rate it is injected. It is possible to know that the CO2 is effectively being adsorbed since the amplitude of the oscillation is smaller than at 25° C. Comparing the pulses at 400° C (curves 1 before and 2 after the heating at 700° C), there is no difference between the first cycle and the second one. There is a much bigger difference between the first and the second pulse sequence at 500° C than at 400° C. This has also been a typical result for La2O3 supported potassium, as will be shown below. This behavior should be originated in surface changes during the treatment at 700oC. The chemistry of La2O3 surface is complex. It is known that different types of carbonates and oxycarbonate species are readily formed under calcination conditions, and that hydroxyl groups are also present. Thus, as discussed below, the decrease in CO2 – surface interaction, is a consequence of a combination of surface transformations that result in a lower basicity. From this point of view, either results obtained with pulses at 400° C or by comparison of the second and following pulses sequences at 500° C should be carried out in order to get information regarding catalyst stability, not including the mentioned changes, such as surface dehydroxylation.

Fig. 3. High Frequency CO2 Pulses with K(4,5 wt. %)/La2O3 catalyst. Four consecutive cycles with intermediate heating steps at 700oC
Figure 3 shows the results obtained with K(4.5)/La2O3 in four consecutive series of pulses at 500, 400° C, and heating up to 700° C between sequences. The promotion with potassium increases the interaction with the CO2, as previously found on MgO and La2O3 and CeO2 (Querini et al,1999 and Miró et al, 1999) using a single pulse technique. The pulses at 500° C again show the very strong interaction between the catalyst and the CO2, a larger number of pulses than in the case of the pure oxide being necessary to approximate the pseudo steady state. After heating up to 700oC (sequence 2), the interaction strongly decreases. Between the second and the fourth pulse sequence, the difference is very small, but still observable. As seen in Fig. 1 the TPO profile of this catalyst calcined at 400° C and at 700° C indicate that the catalytic activity is essentially the same, and that the thermal treatment at 700° C during 2 h does not significantly affect the catalytic performance of this material for the soot combustion reaction. There are two differences between these TPO profiles: the main peak is wider in the case of the catalyst calcined at 700° C, and the second peak is shifted to higher temperatures. The visible change in the level of interaction with CO2 is indicated in the activity test as a minor change. As said before, the XPS results of the catalysts containing 4.5 and 7.5 % K, and calcined at 400 and 700° C (see Table 1) show an increase in the K/La ratio as the calcination temperature increases, in both catalysts, with 4.5 and 7.5 %K. This change in surface composition should also be related to the changes of interaction between the CO2 and the catalyst. The XPS measurements indicate that K/La ratio changes from 0.75 to 0.90 when changing the calcination temperature from 400 to 700° C. The XRD experiments did not show new phases after calcination at 700° C (spectra not shown). The CO2 pulses shown in Fig. 3 also indicate that there is a change in the surface of the catalyst, since the level of interaction between the catalyst and the CO2 is decreasing after each thermal treatment at 700° C. Therefore, even though the activity displays a minor change, the catalyst surface is changing continuously and approaching a stabilization, that was not reached after 4 thermal treatments at 700° C.

Fig. 4. Effect of a pretreatment with SO2 (1000 ppm) at 400oC on the response to High Frequency CO2 Pulses at 500oC (A) and on TPO experiments (B). Catalyst: K(7.5 wt%)/La2O3.
Figure 4A shows the results of pulses sent to the catalyst with 7.5%K and the same catalyst after being treated with air containing 1000 ppm of SO2, at 400° C during 8 h. The catalyst treated with SO2, has a different type of interaction with the CO2 as compared to the untreated catalyst. The first cycle at 500° C is more similar to the pure La2O3 than to the K(7.5)/La2O3. Even though there is a strong interaction, the catalyst almost equilibrates during the 20 pulses of the experiments, while the K(7.5) is still far from this situation. Correspondingly, there is a decrease in the catalytic activity of the catalyst after the treatment with SO2, as shown in Fig. 4B (compare TPO profile of 7.5%K and the same catalyst treated with SO2). Again, a major decrease in the interaction of the catalyst with the CO2 is correlated with a decrease in the activity.
C. Soot-catalyst contacting effects
Figure 5 shows the TPO profiles of mechanical mixtures of catalyst and soot, and when an active phase, like potassium, La2O3 or both were directly deposited on the soot. When the potassium is directly deposited on the soot, the activity is higher than when deposited on a support like SiO2. In this latter case, the pulse experiments (not shown), indicate that there is very low

Fig. 5. Soot-catalyst contacting effect studies. (A) Different solids deposited on soot. (B) K/SiO2 mixed with La2O3 and soot is compared with the mixture K/La2O3 with soot; S: soot; symbol "/" indicates: impregnated on; symbol "+" indicates mechanical mixture.
interaction between the K/SiO2 catalyst and the CO2, therefore indicating that the reaction path through the carbonate-type intermediate would not take place. Correspondingly, the activity of this catalyst is worse than when the active phase, potassium, is directly deposited on the soot. In this case, the contact problem overcomes the catalyst activity, since the comparison is being done between a bad catalyst (K/SiO2) with a bad contact, and a similar catalyst (K) with good contact since it is directly deposited on the soot. A similar situation is found when comparing the La2O3 physically mixed with the soot (not shown), with the case of the oxide deposited directly on the soot. The latter has a better activity. However, if a good catalyst, like K/La2O3 is physically mixed with the soot, the high activity and the synergistic effect between K and the La2O3 overcomes the bad contact with the soot. When both active components are deposited on the soot (K/La2O3/S), the temperature required to burn it is even higher than when any of the active components is deposited alone on the soot. This is not expected, since there should not be a decrease in the activity of K due to the presence of La2O3 or vice versa, and therefore, the activity in the case of the co-impregnated soot should be at least the same as the K impregnated on the soot, since this is the component which is more active when deposited alone. Therefore, there should be a problem during the impregnation step, which leads to this result. The reason of this was not further investigated, and is a demonstration of the difficulties found when trying to form a complex catalyst on the soot surface.
D. Simultaneous removal of soot and NOx on Ba – loaded catalysts
In order to simultaneously abate soot and NO, catalysts containing Ba were prepared. It has previously been reported (Mahzoul et al., 1999) that catalysts containing Pt and BaO are good NO traps, forming nitrates that afterwards decompose. This mechanism is similar to the one proposed above for CO2 forming carbonates that decompose at reaction temperature. Ba catalysts supported in La2O3 and CeO2 were prepared. The Ba content was varied between 10 and 22%, and that of potassium between 4,5 and 7 %. By increasing the Ba contents in Ba/CeO2 catalysts, a slight improvement in activity can be observed. However, these catalysts are less active than those shown in Fig. 1. The maximum in the TPO profile is close to 480° C for Ba(10)/CeO2, and to 430° C for Ba(22)/CeO2 (not shown). This means that even though Ba presents a positive catalytic effect for soot burning, this effect is too small. By adding potassium to these catalysts, an increase in activity can be observed. The Ba(22),K(7)/CeO2 catalyst, calcined at 400° C, presents an activity similar to that of the best catalysts shown in Fig. 1. Figure 6A shows TPO profiles for Ba(22),K(7)/CeO2 catalyst calcined at 400° C, and treated with wet air (saturated with water at room temperature). The treatment was carried out at 400° C for 8, 30, and 100 h. The most noticeable change is that the peaks become a little wider, but the main maximum in the TPO profile occurs essentially at the same temperature. Figure 6B shows results obtained with the Ba(22),K(7)/CeO2 catalyst, but in this case the catalyst was treated with air containing 1000 ppm of SO2. The treatment was carried out also at 400° C, for 30, and 90 h. The TPO profiles shift to very high temperatures, similar to those obtained with the catalyst without potassium. The result obtained after 30 h is very similar to the result obtained after 90 h. It means that the catalyst is deactivated at the beginning of the treatment with SO2. Sulfur is detected by XPS, what suggests the formation of sulfates. Additional experiments and characterizations are being carried out to determine what type of compounds are formed.

Fig. 6. TPO profiles: the effect of water (A) and SO2 (B) on the activity of Ba,K/CeO2 catalyst.
The CO2 pulses carried out on the Ba(22) and Ba(22),K(7) catalysts indicated that the Ba(22)/CeO2 has little interaction with the CO2, and therefore the activity is correspondingly low, as above mentioned.
In order to study the interaction between NOx and the surface of Ba promoted catalysts (Ba/CeO2 and Ba,K/CeO2) gravimetric studies were carried out. Figure 7 displays microbalance experiments, in which we measured the weight increase due to NOx adsorption on Ba/CeO2 catalyst. When only NO (in He) is fed, a small weight increase takes place due to NO adsorption (not shown). However, this effect is reversible when increasing temperature (both with NO in the feed or in pure He atmosphere). When the mixture of NO and O2 was fed (Fig. 7), the weight increase was higher and non-reversible, due to the formation of stable nitrate species. The nitrate formation takes place from the reaction of the NO2 formed and the supported BaO. Similar results (not shown) were obtained with Ba,K/CeO2 catalyst.

Fig. 7. Microbalance results. Gaseous feed: NO and O2 (in He). Catalyst: Ba/CeO2.
Figure 8A shows isothermal soot combustion experiments, with and without NO in the feed. It is observed that lower conversions of soot are obtained when NO is present in the gaseous feed. Even though still under study, we believe that barium nitrate inhibits the activity for soot combustion. Thus, the presence of Ba provides of an efficient nitric oxide trap (as seen in microbalance experiments) but in detriment of soot combustion activity. However, in TPO experiments (Fig. 8B) it can be seen that at temperatures lower than 400oC, the soot conversion is slightly increased when NO is in the feed, probably due to the burning of hydrocarbons adsorbed on soot particles. New formulations are under study with the objective of improving the soot combustion activity of Ba-containing catalysts. An interesting approach would be to obtain a NOx catalytic trap in which the Ba(NO3)2 formed is continuously decomposed to N2 and BaO due to the reducing action of soot particles. To verify this possible reaction pathway, barium nitrate was impregnated on CeO2, and mixed with soot. Then, a TPO profile using helium as carrier gas was carried out.
Figure 9 shows the TPO profiles for this catalyst compared to Ba/CeO. The catalyst containing Ba(NO3)2 is more effective in the soot oxidation due to the action of the NO2 generated in the nitrate decomposition.

Fig. 8. Isothermic (400oC) (A) and Temperature programmed (B) oxidation of soot with Ba/CeO2. Gaseous feed : O2 (6%), NO (0.5%) when included (open symbols) diluted in He.

Fig. 9. Oxidation of soot with supported Ba(NO3)2 and Ba without oxygen in the gaseous feed (pure He flow).
IV. CONCLUSIONS
The support plays an important role in the soot combustion reaction. La2O3 contributes with a reaction path through the formation of carbonate-type intermediates, which decomposes at the reaction temperature, in a similar way to the effect introduced by potassium in all the catalysts. The addition of Co to the catalyst supported on La2O3, to supply the redox way, leads to a loss of the thermal stability as a consequence of the formation of an oxide with perovskite-type structure.
The high frequency CO2 pulse proved to be very useful to obtain information about minor changes in the catalyst surface upon different treatments. In many cases, the changes in the surface are so small, that are not reflected by an activity modification. However, in all cases when the interaction between the catalyst and the CO2 drastically change, there is a correlation between the activity and this level of interaction. This is the case of the catalysts treated with SO2, or when comparing catalyst with and without K.
Ba presents a small catalytic activity for soot burning. The catalyst Ba(22),K(7)/CeO2 has no catalytic effect for NO reaction with soot. However, these catalysts containing Ba and K, supported on CeO2 display good activity and thermal stability. The resistance to wet atmosphere is also good. However, in the presence of sulfur dioxide, the catalyst loses activity very fast. The reason for this is now being studied.
Acknowledgements
The financial support received from ANPCyT (PICT 14-6971) and from Universidad Nacional del Litoral (CAID Program) is acknowledged. Thanks are also given to Matías Céspedes for his contribution with the isothermal runs.
REFERENCES
1. Heck, R.M. and Farrauto, R.J., Catalytic AirPollution Control, Van Nostrad Reinhold ed. (1995). [ Links ]
2. van Setteen, B.A.A.L., Scouten, J.M.,Makee M. and Moulijn, J.A. "Realistic contact for soot with an oxidation catalyst for laboratory studies" Appl.Catal.B: Environmental, 28, 253-257 (2000). [ Links ]
3. Mahzoul, H., Brilhac, J.F. and Gilot, P., "Experimental and mechanistic study of NOx adsorption over NOx trap catalysts" Applied Catalysis B: Environmental 20, 47-55 (1999). [ Links ]
4. Miró, E.E., Ravelli, F., Ulla, M.A., Cornaglia, L.M. and Querini, C.A., "Catalytic combustion of diesel soot on Co,K supported catalysts", Catalysis Today 53, 631-638 (1999). [ Links ]
5. Miró, E.E., Ravelli, F., Ulla, M.A., Cornaglia,L.M. and Querini, C.A., "Catalytic diesel soot elimination on Co-K/La2O3 catalysts: reaction mechanism and the effect of NO addition", Stud. In Surf. Sci. and Catal. 130, 731-736 (2000). [ Links ]
6. Pisarello M.L., Milt V.G., Peralta M.A., Querini C.A. and Miró E.E., "Simultaneous removal of soot and nitrogen oxides from diesel engine exhausts", Catalysis Today 75, 465-470 (2002) [ Links ]
7. Querini, C., Ulla, M.A., Requejo, F., Soria, J., Sedrán, U. and Miró, E., "Catalytic combustion of diesel soot particles. Activity and characterization of Co/MgO and Co,K/MgO catalysts"Appl. Catal.B: Environmental, 15, 5-19 (1998). [ Links ]
8. Querini, C., Cornaglia, L.,Ulla, M.A. and Miró, E., "Catalytic combustion of diesel soot on Co,K/MgO catalysts. Effect of the potassium loading on activity and stability". Appl. Catal. B: Environmental 20, 165-177 (1999). [ Links ]

