










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkLatin American applied research
versión impresa ISSN 0327-0793
Lat. Am. appl. res. v.37 n.4 Bahía Blanca oct. 2007
Preferential oxidation of CO in presence of H2 behavior OF PtSn/γ -Al2O3 catalysts modified by K or Ba
G. J. Siri1,2, G. R. Bertolini1,2 and O. A. Ferretti1,2
1 Centro de Investigación y Desarrollo en Ciencias Aplicadas "Dr. Jorge Ronco" (CINDECA), Facultad de Ciencias Exactas, Universidad Nacional de La Plata-CONICET, 47 N°257, 1900 La Plata, Argentina.
cindeca@unlp.edu.ar
2 Facultad de Ingeniería, Universidad Nacional de La Plata, 1 esq 47, 1900 La Plata, Argentina.
diqs@ing.unlp.edu.ar
Abstract — The behavior of catalysts based on Pt/γ-Al2O3 is analyzed in the CO oxidation reaction. Experiments were performed in the temperature range between 50°C and 250°C with a feed H2 rich, and a concentration of 1% CO and 0.6% O2 Catalysts studied were: monometallic Pt/γ-Al2O3, bimetallic PtSn/γ-Al2O3 with a ratio Sn/Pt=1.6 at/at and two catalysts prepared by modification of the last one with K or Ba. The catalyst that showed maximum activity and selectivity levels is the PtSnK/γ-Al2O3. The tin addition increases the activity which allows the reaction to occur at lower temperatures due to the decrease in the CO-Pt interaction. The addition of K or Ba produces a subsequent improvement with respect to the CO oxidation temperature, which leads to a selectivity increase. The selectivity of the CO oxidation passes through a maximum value as a function of the temperature. This maximum is explained by the importance acquired by the reverse of water gas shift reaction at temperatures near 200ºC.
Keywords — Preferential Oxidation. Hydrogen Purification. PtSn/γ -Al2O3.
I. INTRODUCTION
The requirements imposed world wide demand the search of technological alternatives, although the control of gaseous emissions from conventional thermal machines has experienced some advances. One of these alternatives is the use of fuel cells fed by hydrogen, that allow to optimize the use of the more scarce energetic resources since they are capable to convert the energy from chemical to mechanical one with an efficiency quite higher than the one of the conventional thermal machines. For example, in the case of internal combustion engines of mobile sources, the fuel cells are able to almost duplicate their efficiency with null or minimum emissions of pollutants.
The hydrogen is a fuel with difficulties for its distribution as well as for its storage, consequently the challenge is to generate it in situ (on board) or in a service station, from the reforming or partial oxidation of natural gas, hydrocarbons, methanol or ethanol. In all cases the formation of carbon oxides (CO and CO2) is unavoidable. The carbon monoxide must be eliminated because in concentrations higher than 20 ppm it leads to the deactivation of the electrodes of the fuel cells type PEM (Polymer Electrolyte Membrane) used for these systems (Manasilp and Gulari, 2002)
The need that the generated hydrogen must be an ultra pure product forces to technological solutions such as membrane reactors or catalytic processes for purification. In the case of membrane reactors, that allow the selective passage of hydrogen, one of the difficulties to defeat with actual technologies is the necessity to operate with a high pressure difference on both sides of the membranes (Irusta et al., 2005). In catalytic purification processes part of the carbon monoxide can be eliminated via water gas shift reaction (WGSR), up to levels between 500 and 1000 ppm fixed by thermodynamics. The CO remainder can be eliminated by methanization or by selective oxidation (Preferential oxidation, PROX), in which the conversion of hydrogen must be minimized. The methanization is easily performable in presence of catalysts based on Ni (Takenaka et al., 2004; Schubert et al., 2004; Farrauto, 2005); however, they present as a problem the need to reach high temperature levels for an effective elimination (>250ºC); on the other hand the use of Ni in mobile applications is considered a potential health hazard.
Since investigations about the development of catalysts with higher tolerance to the CO presence continue, the PROX appears as the most adequate way to feed ultra pure hydrogen to the fuel cells. In this case different catalytic systems have been studied and they can be classified in three groups:
(i) Catalysts based on supported Au; they present a very high activity at low temperatures (Margitfalvi et al., 2004; Trimm, 2005). These systems are strongly dependent on the preparation method and in general they present deactivation problems and penalization of the selectivity by hydrogen oxidation at temperatures higher than 80ºC (Recupero et al., 2004). Evidences about resistance to poisoning and to sintering through experiments at prolonged times seem necessary for these systems.
(ii) Catalysts based on supported Cu were also studied with good results for the activity, although problems inherent to Cu with respect to the stability and to the control of its oxidation state seem not to be defeated (Recupero et al., 2004; Shiau et al., 2006; de Souza, et al., 2006).
(iii) Catalysts based on noble metals (Pt, Ru, Rh, Pd) have demonstrated to be active in the oxidation of CO to CO2 even at low temperatures (Roberts et al., 2003; Mariño et al., 2004; Wootsch et al., 2004; Bourane and Bianchi, 2004; Avgouropoulos and Ioannides, 2005). In the case of these type of catalysts modified by tin, it has been demonstrated that the interaction MSn (M:Pt, Pd) increases the oxidizing activity (Grass and Lintz, 1977; Margitfalvi et al., 2002), which is attributed to the presence of SnOx sites in intimate interaction with the noble metal. Furthermore, alkali-metal promotion of platinum and rhodium catalysts was observed to enhance the catalytic performance of these catalysts in preferential oxidation (PROX) of CO in hydrogen-rich streams for fuel-cell applications (Tanaka et al., 2003, Mirkelamoglu and Karakas, 2006).
In this work, the behavior of Pt supported on γ-Al2O3 catalysts, modified by Sn, is analyzed in presence of alkaline or alkaline earth metals (K, Ba). The Sn addition by controlled preparation techniques coming from the surface organometallic chemistry on metals (SOMC/ M) produces geometric and electronic modifications that induce to changes in the catalytic properties of the active phase as it was observed in previous works (Siri, et al., 2005a). The changes by addition of alkaline and alkaline earth metals generate modifications not only on the acid-basic properties of the support but also on the properties of the metallic phase (Siri, et al., 2005b).
II. METHODS
A. Catalyst Preparation.
A commercial γ- Al2O3 (Cyanamid Ketjen) crushed to a size of 60-100 mesh was used as support. For the preparation of the platinum monometallic catalyst, an impregnating solution of H2PtCl6 was added onto the support so as to obtain 1% w/w Pt exchanged. Then, the solids were repeatedly washed, dried at 105 °C, calcined in air at 500 °C and reduced in flowing H2 at the same temperature, leading to the monometallic Pt/ γ- Al2O3 catalyst. After the reduction step, the Pt/ γ- Al2O3 catalyst was washed several times with a NH3 aqueous solution (0.1 M) at room temperature, in order to obtain a chlorine concentration under 0.1% in the resulting solid. The bimetallic systems have been prepared by reaction between the monometallic catalyst and SnBu4 dissolved in n-decane, maintaining a H2 flow of 30 cm3 min-1 and a temperature of 150°C during the reaction time in order to obtain Sn/Pt ratio of 1.6 as it is reported in references (Siri, et al., 2005a). After 4 h of reaction, the systems were cooled to room temperature and were repeatedly washed with n-heptane under a H2 atmosphere. The elimination of the organic groups heating the solids in a H2 atmosphere at 500°C generates the supported bimetallic PtSn catalysts. PtSnM/ γ-Al2O3 (M: Ba, K) were obtained by adding impregnating solutions of Ba(NO3)2 and KOH, so as to obtain M/Pt ratios of 8.5 and 10, respectively. Metallic contents were determined by Atomic Spectrometry.
B. Charaterization.
Transmission Electron Microscopy (TEM). The size distribution of metallic particles was determined by using a Jeol 100 CX instrument. To estimate the mean particle size (dva), the particles were considered spherical and the second moment of the distribution was employed.
X-ray photoelectron spectroscopy (XPS). The samples were re-reduced in situ in the pre-treatment chamber of the spectrometer. Spectra were obtained with a VG ESCALAB 200R spectrometer equipped with a hemispherical electron analyzer and an Mg Kα 120 W X ray source. The reduction treatment was carried out in situ, by heating the samples under a hydrogen flow at 500°C for 1 h. As internal standards the binding energy (BE) of the Al 2p peak at 74.5 eV was considered. The intensities were estimated by calculating the integral of each peak after subtraction of the S-shaped background and fitting the experimental peak to a Lorentzian/ Gaussian mix of variable proportion.
C. Activity Measurements.
The catalytic tests were performed by placing the sample in a conventional flow reactor (6 mm internal diameter, 50 mg of catalyst sample) at atmospheric pressure and 50-250°C. The composition of the reaction products was analyzed on line with the reactor by using a Carlo Erba Fractovap series 2150 gas chromatograph with a FID detector and a Shimadzu GC 8A with TCD detector. Total flow feed was 100 cm3min-1. Feed composition was 1% CO, 0.6% O2, 9% He and 89.4% Ar for tests in hydrogen absence and 1% CO, 0.6% O2, 9% He and 89.4% H2 for oxidation tests in hydrogen presence. Carlo Erba chromatograph was employed to analyze CO and CO2. The effluent of the reactor contains carbon oxides which can be separated in a Porapak 60/80 column of 3 m length and 3 mm internal diameter, kept at 30°C and using H2 as carrier. After separation, CO and CO2, in the presence of hydrogen carrier were methanized in a fixed bed reactor with a Ni/Al2O3 catalyst at 450°C, so as to analyze the two methane peaks.
Shimadzu chromatograph was employed to analyze H2, N2 and O2 with a Molecular Sieve 4A column of 3 m length and 3 mm of internal diameter
D. Results and Discussion
TEM results show a narrow distribution of metallic particle sizes, with a mean particle size of 1.8 nm for monometallic platinum catalyst. This value indicates a high dispersion of the platinum phase (around 60%, Farrauto and Bartholomew, 1997). The particle size distribution of the bimetallic system PtSn/γ- Al2O3 is quite similar to the corresponding monometallic one, presenting a moderated shift (0.4 nm) compatible with a selective deposition of tin over platinum, in agreement with previously reported results involving these kind of preparation procedures (Siri, et al., 2005a; Siri, et al., 1997; Casella et al., 2000). TPR diagrams show two main peaks of hydrogen consumption, in the regions of 75-110ºC and 360-440ºC, assigned to a weak and a strong metal support interaction, respectively (Kappenstein et al., 1995).
XPS results show that platinum is completely reduced in all the samples studied, because the spectra contain only one peak corresponding to Pt centered around 314-315 eV (Pt 4d5/2). The reference value was taken from the NIST X Ray Photoelectron Spectroscopy Database (Wagner, 1989). In PtSn/ γ- Al2O3 system, a downward shift of ca. 1 eV in the binding energy of platinum was observed with respect to the corresponding monometallic catalyst. This fact would be indicative of the existence of an electronic effect of tin over platinum, in agreement with previously published results for PtSn/SiO2 catalysts prepared by Stagg et al. (1997). Studies carried out by Shen et al. (1999) from quantum chemical calculations employing density functional theories (DFT) for Pt19 and Pt16Sn3 clusters indicate that tin donates electrons to the 6sp and 5d orbitals of platinum. Rodríguez et al. (1998 a, b) investigated platinum-tin surface alloys using synchrotron-based high resolution photoemission and ab-initio self consistent field calculations and concluded that Pt-Sn bond involves a Sn (5s, 5p) → Pt (6s, 6p) charge transfer together with a Pt (5d) → Pt (6s, 6p) rehybridization. In this same sense, in a previous study of a PtSn/SiO2 system by Pt L2,3 XANES, the existence of an electronic effect was also explained by means of the d → s, p rehybridization process taking place in PtSn 3-dimensional small nanoclusters leading to an increase in the number of Pt 5d holes (Ramallo López et al., 2003).
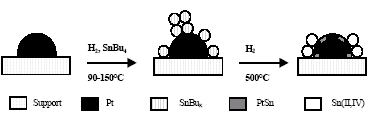
Concerning tin oxidation state, the Sn 3d5/2 peak obtained by XPS contains two contributions. The first with a binding energy of around 484 eV is assigned to Sn(0) according to the well determined values (484.9 eV) obtained by Rodríguez et al. (1998a); the second contribution corresponds to Sn(II, IV), with a binding energy centered around 487 eV (Wagner, 1989). Results obtained for the fraction of metallic tin with respect to total tin (Sn(0)/SnTOTAL) and Sn(0)/Pt ratio are also presented in Table 1. All supported bimetallic catalysts showed Sn(0)/SnTOTAL and Sn(0)/Pt ratios of around 0.2 and 0.35, respectively. In this case, the existence of ionic tin is demonstrated. Its origin may be assigned to the migration of part of the tin, initially deposited onto the platinum via the reaction with SnBu4, to the platinum-support interface where it could be forming aluminate. It is important to mention that blank impregnation experiments of SnBu4 onto γ-Al2O3 demonstrated that in Pt absence there is no reaction between the or ganometallic compound and the support. In relation to Sn(0) atoms, the existence of a PtSn alloy interacting with diluted metallic Pt atoms was shown in a previous paper by using EXAFS experiments for PtSn/ γ- Al2O3 catalysts. The evolution of active phases starting from the monometallic catalyst Pt/ γ- Al2O3 is shown in Scheme 1 (Siri et al., 2005a).
Table 1. TEM and XPS results and denomination of the studied catalysts.

Scheme 1. Representation of catalytic surface for PtSn catalysts.
With respect to catalysts modified by Ba and K, an effect of electronic nature on the Pt is also observed. In this case, this effect is interpreted as a positive shift of 0.3 and 1 eV for Ba and K, respectively. This fact indicates that besides the known modifications that alkaline or alkaline earth metals produce on the acid-basic properties of the support (Siri et al., 2005b), they also interact with the base metal as it appears from the changes observed in its electronic properties.
CO oxidation in hydrogen absence.
In these experiments, catalysts were previously reduced to 500 °C in H2 atmosphere, then the hydrogen was replaced by Ar and the reactor was cooled up to a temperature of 50°C. Just in this moment, the reactive mixture was fed with the following composition: 1% CO, 0.6% O2, 9% He and 89.4% Ar. After two injections at the same conditions, the temperature was increased 50°C until reach a maximum temperature around 250°C. In this way, a precise information about the oxidizing properties with respect to the carbon monoxide is obtained for the different active phases by using a CO/O2 ratio very near to the stoichiometric one. This means to be placed in the most possible severe condition with respect to the oxidation of CO to CO2. Results obtained are shown in Fig. 1, where it is possible to observe that all catalytic systems tested convert completely the CO at temperatures around 200°C.
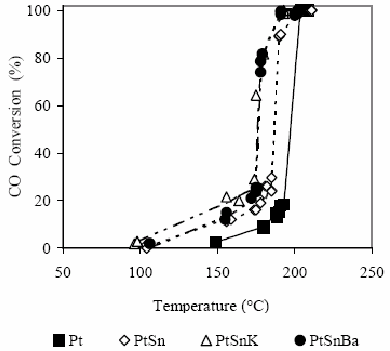
Figure 1. CO conversion as a function of the temperature for the different studied catalysts in H2 absence. For experimental conditions see the text.
Due to the reaction exothermicity, the existence of hot spots must not be discarded reaching in the solid phase temperatures higher than the ones really read by the thermocouple placed in the central sheath of the reactor (Ouyang and Besser, 2005). This situation would allow to explain the nature of the curves observed in this type of selective oxidation reactions of CO (Mariño et al., 2004, Wootsch et al., 2004, Özdemir et al., 2004, Dudfield et al., 2001), where the conversion passes from values of about 15% to values of around 100% in a narrow temperature range; however, it is important to mention that this effect is interpreted by some authors as a change in the reaction mechanism as a function of the temperature (Bourane and Bianchi, 2004).
In Fig. 1, the monometallic catalyst of Pt presents the lowest activity level between the different catalysts tested, reaching a 15% CO conversion at 190°C. The bimetallic catalyst PtSn shows a better oxidation capacity since it reaches a conversion of about 15% at 160°C, while at 190°C, a 100% CO conversion is reached. Bimetallic PtSn catalysts modified with K or Ba are the ones that present the best activity levels achieving conversions of 15% at about 145ºC reaching 100% at around 180°C.
Taking into account these results it is evident that the Sn as well as the K or Ba addition presents a promoter effect from the oxidizing capacity of the base metal (Pt). This is in agreement with that previously shown (Table 1) where it is possible to observe that the Sn addition to catalysts based on Pt modifies geometric and electronic properties of the active phase (Margitfalvi et al., 2002, Akin et al., 2001, Okanishi et al., 2006). According to microcalorimetric studies it was possible to demonstrate that there is a strong diminution of the adsorption heat of the CO by the Sn addition to Pt/SiO2 catalysts (Cortright and Dumesic, 1995), as the oxidation kinetics can be represented by an expression according to a Langmuir-Hinshelwood mechanism, where the CO appears in the denominator of such expression; this decrease of the adsorption heat allows to explain reasonably the increase of the catalytic activity (Akin et al., 2001). On the other hand, the Sn as it arises from XPS results shown in Table 1 is present in important proportions as Sn ionic which confers it the capacity to promote the O2 adsorption, followed by the reverse-spillover from the oxide onto Pt sites and surface reaction occurring on Pt sites between CO adsorbed and O (Grass and Lintz , 1997, Akin et al., 2001).
The promotion of the CO oxidation by the addition of alkaline or alkaline earth metals (K, Ba) is in agreement with results reported in PdO/SnO2 catalysts promoted by Na. In this case the catalyst showed an increase in the oxidizing activity of CO to CO2, explained
by the authors because of the formation of a ternary oxide (with the formula NaxPd3O4) able to increase the O2 storage capacity (Mirkelamoglu and Karakas, 2006).
CO oxidation in hydrogen presence.
In these experiments, catalysts were previously reduced at 500 °C in H2 atmosphere, the reactor was cooled up to 50 °C and then the reactive mixture was fed with the following composition: 1% CO, 0.6% O2, 9% He and 89.4% H2. In this case an approximate stoichiometric ratio of CO/O2 is maintained, for a H2/CO ratio around 90, which represents an extremely severe condition for the analysis of these systems from the point of view of the catalytic process.
Conversion results of CO and O2 are shown in Fig. 2 and 3. In this case, in H2 presence, the difference of the behavior between catalysts with and without Sn is extremely marked. The monometallic catalyst shows that the CO conversion becomes appreciable at 100°C (3%), while the O2 conversion reaches in these conditions the 15%. A 15% CO conversion is observed at 150°C and O2 is completely consumed. At 210°C a CO conversion of about 68% is reached.
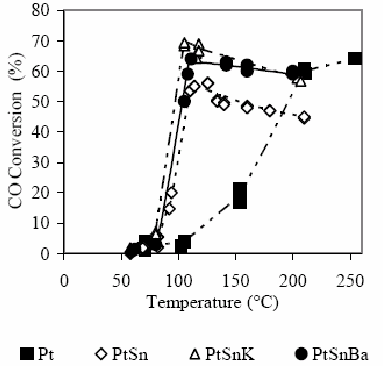
Figure 2. CO conversion as a function of the temperature for the different studied catalysts in H2 presence. For experimental conditions see the text.
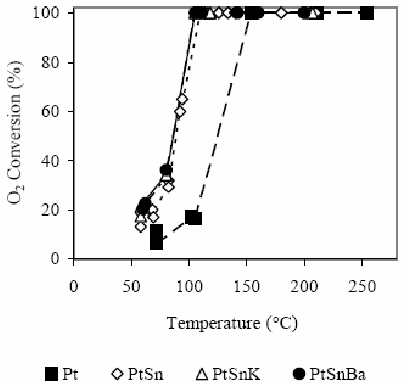
Figure 3 O2 conversion as a function of the temperature for the different studied catalysts in H2 presence. For experimental conditions see the text.
Defining the selectivity for the CO oxidation as the half of the CO2 amount produced in relation to the O2 amount consumed, it is clearly evident that at low temperatures the selectivity is low since the O2 consumption is produced principally by water formation. The bimetallic catalyst PtSn produces the CO oxidation reaction at lower temperature than the monometallic catalyst. The selectivity increases with the temperature increase up to reaching a temperature of about 115°C. The maximum conversion reached is about 55%. A subsequent temperature increase decreases slightly the selectivity as it was also observed by Manasilp and Gulari (2002) and Kahlich et al. (1997). The addition of K or Ba decreases the temperatures of the CO maximum conversion as well as the total O2 consumption. Multimetallic catalysts improve appreciably the activity of the CO oxidation and also the selectivity, as it is shown in Table 2.
Table 2. Maximum selectivities reached for the different studied catalysts. 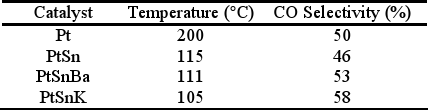
In this case, where the selectivity at low temperatures depends on the relative activities of the oxidation of CO to CO2 and the oxidation of H2 to H2O, the mentioned effect, due to modifications in the CO adsorption in PtSn systems with respect to supported Pt, produces a selectivity increase. The decrease of the chemisorptive properties of both reagents influences on the reaction. On one hand, the decrease in the CO adsorption allows a higher reaction rate toward the CO2 production due to a weakening of the strong interaction CO-Pt that originates the inhibition of the active sites (the global reaction order for the CO is negative). On the other hand, the decrease of the H2 adsorption decreases the formation rate of water while the amount of active sites covered by this reagent diminishes.
In catalytic tests, the selectivity passes through a maximum with the temperature increase as it was observed in references (Manasilp and Gulari, 2002; Kahlich et al., 1997). This analysis is quite complex and subject to interpretations that sometimes could be questionable. Generally, the occurrence of thermal effects produced by the strong exothermicity of involved reactions is not taken into account (reaction heats of CO to CO2=-2.83x105 J/mol and of H2 to H2O=-2.42x105J/mol, Ouyang and Besser, 2005). These thermal effects could explain the light off observed in Fig. 1, 2 and 3 (conversion vs. T), where it is clear that the slope of curves is lower in the monometallic Pt catalyst than in PtSn and PtSnK catalysts. These results are coherent with the higher oxidizing activity observed in multimetallic catalysts with respect to the monometallic one, which has a stronger Pt-CO interaction.
With conditions used in these tests, the reverse of water gas shift (RWGS) reaction must be taken into account (CO2+ H2↔ CO + H2O ) . This reaction has been largely studied in homogeneous and heterogeneous catalysis associated in general to reactions that generate hydrogen or syngas, where it is accepted that the water gas shift reaction is in thermodynamic equilibrium (Wei and Iglesia, 2004). Taking this into account, the equilibrium constant was calculated from the exit compositions of the reactor (KeqCalc); with this value the temperature at which the thermodynamic equilibrium constant is equal at KeqCalc was determined (TCalc). In Table 3, the temperature measured in the central sheath of the reactor (TReact) and the estimated temperature (TCalc) was compared. The estimated temperature, TCalc , would be an indicative of the actual temperature on the solid phase.
Table 3. Values of the KeqCalc, TCalc, TReact and ΔT=TCalc-TReact
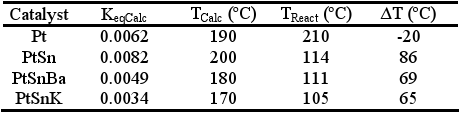
Table 3 shows for modified systems that, the estimated temperature is about 200°C, between 65-85°C, higher than the experimental value. Taking into account that the reverse of water gas shift reaction is found in almost thermodynamic equilibrium conditions, it is not surprising that the CO concentration remains about 0.5% and decreases as the temperature raises. For this reason, the search for systems reaching a high conversion of CO at low temperatures is very important to improve the global yields of this process. In our case, the best activities observed in bimetallic and multimetallic systems could be partially penalized by the RWGS reaction conversion assuming that the calculated reactor temperature (TCalc) gives a more real approximation of the temperature of the catalytic surface.
III. CONCLUSIONS
The catalysts prepared for this work showed to be active for the CO oxidation in the presence of high H2 concentrations at temperatures of about 200°C or lower.
The catalyst that showing maximum activity and selectivity is the PtSnK/ γ-Al2O3. Addition of tin improves the activity and allows the reaction to occur at lower temperatures by a decrease in the CO-Pt interaction. The K or Ba addition produces a subsequent improvement with regard to the CO oxidation temperature, leading to a selectivity increase in the CO oxidation with respect to the H2 oxidation.
The CO selectivity passes through a maximum value as a function of the temperature. This maximum is explained by the importance gained by the reverse of water gas shift reaction at temperatures near 200ºC, which is potentiated by thermal effects not always reported. The optimization of this process must take into account the search for catalytic systems that work at low temperature in order to avoid the influence of the RWGS reaction.
REFERENCES
1. Akin, A.N., G. Kilaz, A.I. Isli and Z.I. Onsan, "Development and characterization of Pt-SnO2/γ-Al2O3 catalysts", Chem. Engn. Sci., 56, 881-888 (2001). [ Links ]
2. Avgouropoulos, G. and T. Ioannides, "CO tolerance of Pt and Rh catalysts: effect of CO in the gas-phase oxidation of H2 over Pt and Rh supported catalysts", Appl. Catal. B: Env., 56, 77-86 (2005). [ Links ]
3. Bourane, A. and D. Bianchi, "Oxidation of CO on a Pt/Al2O3 catalyst: from the surface elementary steps to light-off tests V. Experimental and kinetic model for light-off tests in excess of O2", J. Catal., 222, 499-510 (2004). [ Links ]
4. Casella, M.L., G.J. Siri, G.F. Santori, O.A. Ferretti and M. Ramírez de Agudelo "Surface characterization of Li modified PtSn catalysts for isobutane dehydrogenation", Langmuir, 13, 5639-5643 (2000). [ Links ]
5. Cortright, R.D. and J.A. Dumesic, "Effects of potassium on silica-supported Pt and Pt/Sn catalyst for isobutan dehydrogenation", J. Catal., 157, 576-583 (1995). [ Links ]
6. de Souza, T.R.O., A.J.S. Mascarenhasb and H.M.C. Andrade, "Selective catalytic oxidation of CO in H2", React. Kin. Catal. Lett., 87, 3-9 (2006). [ Links ]
7. Dudfield, C.D., R. Chen and P.L. Adcock, "A carbon monoxide PROX reactor for PEM fuel cell automotive application", Int. J. of Hydrogen Energy, 26, 763-775 (2001). [ Links ]
8. Farrauto, R. J., "Introduction to solid polymer membrane fuel cells and reforming natural gas for production of hydrogen", Appl Catal B: Env., 56, 3-7 (2005). [ Links ]
9. Farrauto, R.J. and C.H.Bartholomew, Fundamentals of Industrial catalytic Processes Ed. Blackie Academic & Professional (1997). [ Links ]
10. Grass, K. and H.G. Lintz, "The Kinetics of Carbon Monoxide Oxidation on Tin(IV) Oxide Supported Platinum Catalysts", J. Catal., 172, 446-452 (1997). [ Links ]
11. Irusta, S., J. Múnera, C. Carrara, E.A. Lombardo and L.M. Cornaglia, "A stable, novel catalyst improves hydrogen production in a membrane reactor", Appl. Catal. A: Gen., 287, 147-158 (2005). [ Links ]
12. Kahlich, M.J., H.A. Gasteiger and R.J. Behm "Kinetics of the Selective CO Oxidation in H2-Rich Gas on Pt/Al2O3", J. Catal., 171, 93-105 (1997). [ Links ]
13. Kappenstein, C., M. Saouabé, M. Guérin, P. Marecot, I. Uszkurat and Z. Paál, "Characterization and activity of Pt-Sn/A12O3 catalysts of different preparation: coimpregnation and new Pt-Sn precursor", Catal. Lett., 31, 9-17 (1995). [ Links ]
14. Manasilp, A. and E. Gulari, "Selective CO oxidation over Pt/alumina catalysts for fuel cell applications", Appl. Catal. B: Env., 37, 17-25 (2002). [ Links ]
15. Margitfalvi, J. L., M. Hegedus, A. Szegedi and I. Sajó, "Modification of Au/MgO catalysts used in low temperature CO oxidation with Mn and Fe", Appl.Catal. A: Gen., 282, 87-97 (2004). [ Links ]
16. Margitfalvi, J.L., I. Borbáth, M. Hegedus, Á. Szegedi, K. Lázár, S. Göbölös and S. Kristyán, "Low temperature oxidation of CO over tin-modified Pt/SiO2 catalysts", Catalysis Today, 73, 343-353 (2002). [ Links ]
17. Mariño, F., C. Descorme and D. Duprez, "Noble metal catalysts for the preferential oxidation of carbon monoxide in the presence of hydrogen (PROX)", Appl. Catal. B: Env., 54, 59-66 (2004). [ Links ]
18. Mirkelamoglu, B. and G. Karakas, "The role of alkali-metal promotion on CO oxidation over PdO/SnO2 catalysts", Appl. Catal. A: Gen., 299, 84-94. (2006). [ Links ]
19. Okanishi, T., T.Matsui, T.Takeguchi, R.Kikuchi and K. Eguchi, "Chemical interaction between Pt and SnO2 and influence on adsorptive properties of CO", Appl. Catal. A: Gen., 298, 181-187 (2006). [ Links ]
20. Ouyang, X. and R.S. Besser "Effect of reactor heat transfer limitations on CO preferential oxidation", Chem. Journal of Power Sources, 141, 39-46 (2005). [ Links ]
21. Özdemir, C., A.N. Akin and R. Yildirim, "Low temperature CO oxidation in hydrogen rich streams on Pt-SnO2/Al2O3 catalyst using Taguchi method.", Appl. Catal. A: Gen., 258, 145-152 (2004). [ Links ]
22. Ramallo López, J.M. , G.F. Santori, L. Giovanetti, M.L. Casella, O.A. Ferretti and F.G. Requejo, "PS and XANES Pt L2,3-edge studies of dispersed metallic Pt and PtSn clusters on SiO2 obtained by organometallic síntesis: structural and electronic characteristics", J. Phys. Chem. B, 107, 11441-11451 (2003). [ Links ]
23. Recupero, V., L. Pino, M. Cordaro, A. Vita, F. Cipitiand and M. Lagana, "CO clean-up transient device integrated to a preferential oxidation reactor for PEFC electric vehicles", Fuel Processing Technology, 85, 1445-1452 (2004). [ Links ]
24. Roberts, G.W., P. Chin, X. Sun1 and J.J. Spivey "Preferential oxidation of carbon monoxide with Pt/Fe monolithic catalysts: interactions between external transport and the reverse water-gas-shift reaction", Appl. Catal. B: Env., 46, 601-611 (2003). [ Links ]
25. Rodriguez, J.A., S.Chaturvedi, T.Jirsak and J.Hrbek, "Reaction of S2 and H2S with Sn/Pt[111] surface alloys: Effects of metal-metal bonding on reactivity towards sulfur", J. Chem. Phys., 109, 4052-4062 (1998a). [ Links ]
26. Rodriguez, J.A., T. Jirsak, S. Chaturvedi and J. Hrbek, "Surface Chemistry of SO2 on Sn and Sn/Pt(111) Alloys: Effects of Metal-Metal Bonding on Reactivity toward Sulfur", J. Am. Chem. Soc., 120, 11149-11157 (1998b). [ Links ]
27. Schubert, M.M., A. Venugopal, M. J. Kahlich, V. Plzak and R.J. Behm, "Influence of H2O and CO2 on the selective CO oxidation in H2-rich gases over Au/α-Fe2O3", J. Catal., 222, 32-40 (2004). [ Links ]
28. Shen, J., J.M. Hill, R.M. Watwe, B.E. Spiewak and J.A. Dumesic, "Microcalorimetric, Infrared Spectroscopic, and DFT Studies of Ethylene Adsorption on Pt/SiO2 and Pt-Sn/SiO2 Catalysts", J. Phys. Chem. B, 103, 3923-3934 (1999). [ Links ]
29. Shiau, C., M.W. Ma and C.S. Chuang, "CO oxidation over CeO2-promoted Cu/γ-Al2O3 catalyst: Effect of preparation method", Appl. Catal. A: Gen., 301, 89-95 (2006). [ Links ]
30. Siri, G.J., J.M. Ramallo López, M.L. Casella, J.L.G. Fierro, F.G. Requejo and O.A. Ferretti, "XPS and EXAFS study of supported PtSn catalysts obtained by surface organometallic chemistry on metals. Application to the isobutane dehydrogenation", Appl. Catal. A: Gen., 278, 239-249 (2005a). [ Links ]
31. Siri, G.J., G.R. Bertolini, M.L. Casella and O.A. Ferretti, "PtSn/ γ-Al2O3 isobutane dehydrogenation catalysts: the effect of alkaline metals addition", Mat. Lett., 59, 2319-2324 (2005b). [ Links ]
32. Siri, G.J., M.L.Casella, G.F.Santori and O.A.Ferretti, "Tin/platinum on alumina as catalyst for dehydrogenation of isobutane. Influence of the preparation procedure and of the addition of lithium on the catalytic properties of the catalyst", IECR 36 4821-4826 (1997). [ Links ]
33. Stagg, S.M., C.A. Querini, W.E. Alvarez and D.E. Resasco "Isobutane Dehydrogenation on Pt-Sn/SiO2 Catalysts: Effect of Preparation Variables and Regeneration Treatments", J .Catal., 168, 75-94 (1997). [ Links ]
34. Takenaka, S., T. Shimizu and K. Otsuda, "Complete removal of CO in hydrogen-rich gas stream through methanation over supported metal catalysts", Int. J. Hydrogen Energy, 20, 1065-1073 (2004). [ Links ]
35. Tanaka, H., S. Ito, S. Kameoka, K. Tomishige and K. Kunimori, "Promoting effect of potassium in selective oxidation of CO in hydrogen-rich stream on Rh catalysts", Catal Communication, 4, 1-4 (2003). [ Links ]
36. Trimm, D., "Minimization of CO in a H2 stream for fuel cell application", Appl. Catal A: Gen., 296, 1-11 (2005). [ Links ]
37. Wagner, C.D., NIST X-Ray Photoelectron Spectroscopy Database, Gathersburg (1989). [ Links ]
38. Wei, J. and E. Iglesia, "Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts", J. Catal., 224, 370-383 (2004). [ Links ]
39. Wootsch, A., C. Descorme and D. Duprez, "Preferential oxidation of carbon monoxide in the presence of hydrogen(PROX) over ceria-zirconia and alumina-supported Pt catalysts", J. Catal., 225, 259-266 (2004).
[ Links ]
Received: May 12, 2006
Accepted: March 20, 2007
Recommended by Subject Editor: Ana Lea Cukierman

